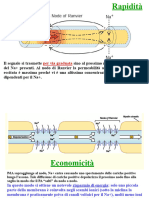
Potrebbero piacerti anche
- Dieta MorroneDocumento12 pagineDieta MorroneVoodoo56% (18)
- Sinapsi e NeurotrasmettitoriDocumento45 pagineSinapsi e NeurotrasmettitoriNicola Laurora0% (1)
- Sistema NervosoDocumento15 pagineSistema NervosoBruce WiiNessuna valutazione finora
- L'impulso NervosoDocumento7 pagineL'impulso NervosoGiancarlo AttivissimoNessuna valutazione finora
- Trasmissione Sinaptica e Integrazione NeuronaleDocumento11 pagineTrasmissione Sinaptica e Integrazione NeuronaleAnnamaria MaderaNessuna valutazione finora
- Fisiologia ValenteDocumento4 pagineFisiologia ValenteFrancesco FalconieriNessuna valutazione finora
- CITOLOGIA E ISTOLOGIA - Lezione 7Documento2 pagineCITOLOGIA E ISTOLOGIA - Lezione 7Federico TosiniNessuna valutazione finora
- Lezione 10 SinapsiDocumento69 pagineLezione 10 SinapsiCopisteria Le PapierNessuna valutazione finora
- Lez9-Miastenia Gravis, PolimiositeDocumento8 pagineLez9-Miastenia Gravis, PolimiositePatty De SimoneNessuna valutazione finora
- Trasmissione SinapticaDocumento12 pagineTrasmissione Sinapticaapi-3774792100% (1)
- Conduzione NervosaDocumento10 pagineConduzione NervosaMiriam LongobardiNessuna valutazione finora
- 7 - Neuroni, Integrazione, NeuromodulazioneDocumento12 pagine7 - Neuroni, Integrazione, Neuromodulazioneapi-26155364Nessuna valutazione finora
- Teoria SinapticaDocumento5 pagineTeoria SinapticaFran BisoNessuna valutazione finora
- 1 AprDocumento35 pagine1 Apralison.pedrini98Nessuna valutazione finora
- Accoppiamento Eccitazione-Contrazione Nel CuoreDocumento35 pagineAccoppiamento Eccitazione-Contrazione Nel CuoreLalla BozziNessuna valutazione finora
- Appunti Di NeurofisiologiaDocumento91 pagineAppunti Di NeurofisiologiaSaraNessuna valutazione finora
- Neuroni NeuromodulazioneDocumento10 pagineNeuroni Neuromodulazioneapi-3774792Nessuna valutazione finora
- Regolazione Dell Attivita CardiacaDocumento54 pagineRegolazione Dell Attivita CardiacaMarco BaldelliNessuna valutazione finora
- S NervosoDocumento104 pagineS NervosoAgostino InzerilloNessuna valutazione finora
- Neuropatie Presentazione 2021 Diana Ele Ultima VersDocumento65 pagineNeuropatie Presentazione 2021 Diana Ele Ultima Versdiana gesinoNessuna valutazione finora
- 2.impulso NervosoDocumento10 pagine2.impulso NervosofNessuna valutazione finora
- Marieb Sintesi 62091 c14Documento3 pagineMarieb Sintesi 62091 c14Caterina NuzzoNessuna valutazione finora
- Il Sistema NervosoDocumento6 pagineIl Sistema NervosoMartina MusmeciNessuna valutazione finora
- BiologiaDocumento1 paginaBiologiaGiulioNessuna valutazione finora
- SDR ParaneoDocumento10 pagineSDR ParaneodenisdeniNessuna valutazione finora
- Intro Eeg BciDocumento28 pagineIntro Eeg BciHibino TsubakiNessuna valutazione finora
- AAA Elettroterapia PDFDocumento51 pagineAAA Elettroterapia PDFOsteofisioNessuna valutazione finora
- (Med ITA) Fisiologia - Automatismo CardiacoDocumento6 pagine(Med ITA) Fisiologia - Automatismo CardiacoZorayda PatriciaNessuna valutazione finora
- 2021 FARMACIA Istologia 7 Tessuto Nervoso 16 MarzoDocumento68 pagine2021 FARMACIA Istologia 7 Tessuto Nervoso 16 MarzoChiara ArdauNessuna valutazione finora
- Fisiologia Generale 5Documento46 pagineFisiologia Generale 5OrlandoCialliNessuna valutazione finora
- Dispensa Fisiologia UnitoDocumento497 pagineDispensa Fisiologia Unitodorian88Nessuna valutazione finora
- Scienze - Il Sistema NervosoDocumento27 pagineScienze - Il Sistema NervosoRiccardo PengoNessuna valutazione finora
- Muscolo Schel - Parte1Documento34 pagineMuscolo Schel - Parte1Francesco MalavoltaNessuna valutazione finora
- Tessuto NervosoDocumento7 pagineTessuto NervosoeristelaNessuna valutazione finora
- Potenziale D'azione CardiacoDocumento53 paginePotenziale D'azione CardiacoLalla BozziNessuna valutazione finora
- Pot CardiaciDocumento47 paginePot CardiaciRiccardo Mason TroiaNessuna valutazione finora
- Lezione 1 Contrazione Muscolare PDFDocumento75 pagineLezione 1 Contrazione Muscolare PDFXXXXXXXXNessuna valutazione finora
- Conduzione Nervosa (Riassunto)Documento10 pagineConduzione Nervosa (Riassunto)Aaron RomanoNessuna valutazione finora
- 3-Farmacologia Del Neurone e Sistema Adrenergico PDFDocumento52 pagine3-Farmacologia Del Neurone e Sistema Adrenergico PDFkarinwarais0% (1)
- Anestetici LocaliDocumento10 pagineAnestetici LocaliChiaraPetraroliNessuna valutazione finora
- Occhio3 Recettori Corteccia VieDocumento43 pagineOcchio3 Recettori Corteccia Viesimo_pNessuna valutazione finora
- Il Sistema NervosoDocumento8 pagineIl Sistema NervosoMartina RossiNessuna valutazione finora
- Domande PIT FisiologiaDocumento5 pagineDomande PIT FisiologiaJoshuaKKyleNessuna valutazione finora
- Cap 4 Riassunto FinDocumento13 pagineCap 4 Riassunto Finandrorru.stefanoNessuna valutazione finora
- 1 Potenziale Di Equilibrio Di NernstDocumento5 pagine1 Potenziale Di Equilibrio Di NernstClaudia OrlannoNessuna valutazione finora
- I Recettori SensorialiDocumento37 pagineI Recettori SensorialiMaryna100% (1)
- NEUROBIOLOGIA La SapienzaDocumento30 pagineNEUROBIOLOGIA La Sapienzachiara cimeiNessuna valutazione finora
- Destefani 1Documento6 pagineDestefani 1MónicaAndreaBarreraNessuna valutazione finora
- Sistema Nervoso - 24 Maggio 2023Documento34 pagineSistema Nervoso - 24 Maggio 2023Gianluca TortoraNessuna valutazione finora
- Pubblicazioni Sulle MicrocorrentiDocumento13 paginePubblicazioni Sulle Microcorrenticarne23Nessuna valutazione finora
- Conduzione Impulso NervosoDocumento32 pagineConduzione Impulso NervosoMarcoNessuna valutazione finora
- 4) Il Sistema Nervoso. Le Cellule Del Sistema Nervoso. Il Potenziale D'azioneDocumento43 pagine4) Il Sistema Nervoso. Le Cellule Del Sistema Nervoso. Il Potenziale D'azionedavide di lorenzoNessuna valutazione finora
- Tessuto NervosoDocumento1 paginaTessuto NervososofiaNessuna valutazione finora
- Occhio Anatomia (Italiano)Documento49 pagineOcchio Anatomia (Italiano)Giulia De SantisNessuna valutazione finora
- Attività Elettrica Del CuoreDocumento26 pagineAttività Elettrica Del CuoreStella FilanteNessuna valutazione finora
- Lezione 10-FisiologiaDocumento5 pagineLezione 10-FisiologiaSarah ScuderiNessuna valutazione finora
- Fisiologia Apparato Cardiocircolatorio 1Documento10 pagineFisiologia Apparato Cardiocircolatorio 1Alessandro Riggi0% (1)
- 8 CervellettoDocumento44 pagine8 CervellettoMoussa LandryNessuna valutazione finora
- MiorisoluzioneDocumento2 pagineMiorisoluzionearabia26Nessuna valutazione finora
- Lo stress ossidativo induce la β-secretasi agendo sull’attività della γ-secretasi: vie di segnale coinvolte e ruolo nella patogenesi della malattia di AlzheimerDa EverandLo stress ossidativo induce la β-secretasi agendo sull’attività della γ-secretasi: vie di segnale coinvolte e ruolo nella patogenesi della malattia di AlzheimerNessuna valutazione finora
- Nanotecnologie per il trattamento dei tumori: quali sono gli effetti sul genoma?Da EverandNanotecnologie per il trattamento dei tumori: quali sono gli effetti sul genoma?Nessuna valutazione finora
- 04 Santoro PlessopatieDocumento53 pagine04 Santoro PlessopatiePietro BracagliaNessuna valutazione finora
- Eset Nod32 Antivirus 13 ItaDocumento141 pagineEset Nod32 Antivirus 13 ItaPietro BracagliaNessuna valutazione finora
- Contratto Quadra FreeDocumento6 pagineContratto Quadra FreePietro BracagliaNessuna valutazione finora
- Decreto Appropriatezza GU n.15 Del 20-1-2016Documento7 pagineDecreto Appropriatezza GU n.15 Del 20-1-2016Pietro BracagliaNessuna valutazione finora
- CNS-Guida Installazione ArubaDocumento17 pagineCNS-Guida Installazione ArubaPietro BracagliaNessuna valutazione finora
- Decreto Appropriatezza - Allegato 2Documento16 pagineDecreto Appropriatezza - Allegato 2Pietro BracagliaNessuna valutazione finora
- Corso Fad Parkinson Strategie TerapeuticheDocumento18 pagineCorso Fad Parkinson Strategie TerapeutichePietro BracagliaNessuna valutazione finora
- Nocicezione e Dolore (Escl Uso Interno)Documento85 pagineNocicezione e Dolore (Escl Uso Interno)Pietro BracagliaNessuna valutazione finora
- Scheda Di Valutazione DoloreDocumento1 paginaScheda Di Valutazione DolorePietro BracagliaNessuna valutazione finora
- Manuale Classeviva Docenti v2.4Documento22 pagineManuale Classeviva Docenti v2.4Pietro BracagliaNessuna valutazione finora
- Bestiario Del Diavolo. Esegesi Biblica Nelle Formulae Spiritalis Intellegentiae Di Eucherio Di LioneDocumento21 pagineBestiario Del Diavolo. Esegesi Biblica Nelle Formulae Spiritalis Intellegentiae Di Eucherio Di LioneLycophronNessuna valutazione finora
- Nastro Di MobiusDocumento19 pagineNastro Di MobiusGiulia MarcelliniNessuna valutazione finora
- Sono Gli Uomini A Fare La StoriaDocumento12 pagineSono Gli Uomini A Fare La StoriaMariarosa La FeminaNessuna valutazione finora
- Legislazione Scolastica - Piccola Dispensa Ad Integrazione Dei Materiali1 PDFDocumento44 pagineLegislazione Scolastica - Piccola Dispensa Ad Integrazione Dei Materiali1 PDFAlessandro SaracinoNessuna valutazione finora
- Il PerdonoDocumento5 pagineIl PerdonoSimone CristinaNessuna valutazione finora
- 20102011ITADocumento11 pagine20102011ITAedunet_italiaNessuna valutazione finora
- Lezione 10 - UstioniDocumento50 pagineLezione 10 - UstionilucaNessuna valutazione finora