Potrebbero piacerti anche
- Seminario 4 Epidemiología - Pruebas Diagnosticas y Puntuales (Corregido Con 1 Audio)Documento18 pagineSeminario 4 Epidemiología - Pruebas Diagnosticas y Puntuales (Corregido Con 1 Audio)Piero EstelaNessuna valutazione finora
- PCI - Espástica: Diagnóstico y tratamientoDocumento44 paginePCI - Espástica: Diagnóstico y tratamientosairaNessuna valutazione finora
- Radiología: Estudio Radiologico de La Articulación de CaderaDocumento49 pagineRadiología: Estudio Radiologico de La Articulación de CaderaDenishitop Lopez JimenezNessuna valutazione finora
- Radiología: Estudio Radiologico de La Articulación de CaderaDocumento49 pagineRadiología: Estudio Radiologico de La Articulación de CaderaDenishitop Lopez JimenezNessuna valutazione finora
- Radiología: Estudio Radiologico de La Articulación de CaderaDocumento49 pagineRadiología: Estudio Radiologico de La Articulación de CaderaDenishitop Lopez JimenezNessuna valutazione finora
- Distrofias Musculares. ClinicaDocumento13 pagineDistrofias Musculares. ClinicaViqui BlancoNessuna valutazione finora
- DISTROFIA MUSCULAR DE DUCHENNE - PPTM PDFDocumento27 pagineDISTROFIA MUSCULAR DE DUCHENNE - PPTM PDFPaola MedinaNessuna valutazione finora
- HipotoníaDocumento112 pagineHipotoníaLucero Auriestela Rivera HernandezNessuna valutazione finora
- Enfermedades NeuromuscularesDocumento8 pagineEnfermedades NeuromuscularesCONSTANZA JAVIERA DÍAZNessuna valutazione finora
- Paralisis Facial y HipotoniasDocumento46 pagineParalisis Facial y HipotoniasNati LuchaNessuna valutazione finora
- Clase 6Documento30 pagineClase 6Mayra Nuria Rojas RojasNessuna valutazione finora
- Síndrome hipotónico: causas, signos y estudio etiológicoDocumento64 pagineSíndrome hipotónico: causas, signos y estudio etiológicoJaime Toro ErbettaNessuna valutazione finora
- Parálisis CerebralDocumento60 pagineParálisis CerebralAlejandra Alcayaga Hidalgo100% (1)
- Trastornos NeuromotoresDocumento35 pagineTrastornos NeuromotoresAlma Solis100% (15)
- HYPERTONIADocumento22 pagineHYPERTONIAXxThekingxXNessuna valutazione finora
- Paralisis CerebralDocumento38 pagineParalisis CerebralIngrid PerezNessuna valutazione finora
- AmitrofiaDocumento21 pagineAmitrofiaarly masielNessuna valutazione finora
- Retraso Madurativo Pensar en DuchenneDocumento40 pagineRetraso Madurativo Pensar en DuchenneSofi C. LugoNessuna valutazione finora
- FibromialgiaDocumento30 pagineFibromialgialocoproanimal100% (1)
- Distrofia Miotónica de SteinertDocumento13 pagineDistrofia Miotónica de SteinertViany RCNessuna valutazione finora
- Paralisis CerebralDocumento12 pagineParalisis Cerebralkarina tosorattiNessuna valutazione finora
- Atrofia Muscular Espinal-1Documento18 pagineAtrofia Muscular Espinal-1Lupita MendozaNessuna valutazione finora
- Detalles y Clasificación de La Atrofia Muscular EspinalDocumento14 pagineDetalles y Clasificación de La Atrofia Muscular EspinalNicolás Daniel BustosNessuna valutazione finora
- Síndrome de Joubert - Signo Del MalarDocumento15 pagineSíndrome de Joubert - Signo Del MalarIvan SantiagoNessuna valutazione finora
- Taller PatologíaDocumento24 pagineTaller PatologíaAlexis GordonNessuna valutazione finora
- Miastenia GravisDocumento33 pagineMiastenia Gravisabraham hernandez cordovaNessuna valutazione finora
- Grupo 11. Resumen Medicina Física y RehabilitaciónDocumento24 pagineGrupo 11. Resumen Medicina Física y RehabilitaciónYiselina RosarioNessuna valutazione finora
- DiabetesDocumento33 pagineDiabetesMarcielys MendozaNessuna valutazione finora
- Ecne - NeuroDocumento7 pagineEcne - NeuroViqui BlancoNessuna valutazione finora
- Clase 4 Paralisis Cerebral InfantilDocumento39 pagineClase 4 Paralisis Cerebral InfantilESTEFANNY ARIASNessuna valutazione finora
- PCI DISTONICAxDocumento22 paginePCI DISTONICAxEhsthephyElizhabethANessuna valutazione finora
- Sindrome de Joubert ExposicionDocumento10 pagineSindrome de Joubert ExposicionHans Edwards CostasNessuna valutazione finora
- Síndromes hipotónicos en lactantesDocumento58 pagineSíndromes hipotónicos en lactantesLourdes Berenice Garfias SorianoNessuna valutazione finora
- Aspectos Clínicos de Las Enfermedades NeuromuscularesDocumento11 pagineAspectos Clínicos de Las Enfermedades NeuromuscularesNATALIA MARTINEZ CORDOBANessuna valutazione finora
- 51 Hipotonia NeonatalDocumento8 pagine51 Hipotonia NeonatalJulio Rolando Pozo CifuentesNessuna valutazione finora
- Abordaje Del Lactante Con Alteraciones Del Tono MuscularDocumento4 pagineAbordaje Del Lactante Con Alteraciones Del Tono MuscularxavittoxNessuna valutazione finora
- Atrofia Medular InfantilDocumento19 pagineAtrofia Medular Infantilchinito90398Nessuna valutazione finora
- Psicomotor Semana 3Documento7 paginePsicomotor Semana 3Angelica AvNessuna valutazione finora
- EncefalopatiaCronicanoevolutiva PDFDocumento16 pagineEncefalopatiaCronicanoevolutiva PDFLujan FernandezNessuna valutazione finora
- Evaluacion Prescripcio AFDocumento74 pagineEvaluacion Prescripcio AFarquitecniaNessuna valutazione finora
- Enfoque Del Paciente Con MiopatiaDocumento40 pagineEnfoque Del Paciente Con MiopatiaFederico Fabio RamosNessuna valutazione finora
- Síndrome Hipotónico PDFDocumento42 pagineSíndrome Hipotónico PDFSaul Yerena100% (3)
- Beneficios de la equinoterapia en personas con síndrome de DownDocumento56 pagineBeneficios de la equinoterapia en personas con síndrome de DownJuanaNessuna valutazione finora
- Caracteristicas de La Distrofia MuscularDocumento7 pagineCaracteristicas de La Distrofia MuscularMartin Guillen HerreraNessuna valutazione finora
- MiopatiasDocumento6 pagineMiopatiasYoltic AlcantaraNessuna valutazione finora
- Caso Clínico - Ataxia de FriedreichDocumento17 pagineCaso Clínico - Ataxia de FriedreichMishel MochaNessuna valutazione finora
- Paralisis Cerebral PDFDocumento48 pagineParalisis Cerebral PDFJosefa Cerda HerreraNessuna valutazione finora
- Medicina Ocupacional Y Rehabilitación: Dra. Blanca R. Serrano GarcíaDocumento50 pagineMedicina Ocupacional Y Rehabilitación: Dra. Blanca R. Serrano GarcíaRosa ArrascueNessuna valutazione finora
- Distrofia MuscularDocumento3 pagineDistrofia MuscularPabloNessuna valutazione finora
- DISTROFIAS MUSCULARES EsgtDocumento4 pagineDISTROFIAS MUSCULARES EsgtJardin Idalis Diaz PayaresNessuna valutazione finora
- Enfermedades MuscularesDocumento74 pagineEnfermedades MuscularesMaría Guadalupe De FriasNessuna valutazione finora
- Temario Hoi 2015Documento282 pagineTemario Hoi 2015Maria Victoria Torres SalazarNessuna valutazione finora
- Distrofia Miotónica de SteinertDocumento13 pagineDistrofia Miotónica de SteinertViany RCNessuna valutazione finora
- Reviisón Bibliográfica - Atrofia Muscular EspinalDocumento6 pagineReviisón Bibliográfica - Atrofia Muscular EspinalbaldovereNessuna valutazione finora
- Clase FibromialgíaDocumento63 pagineClase FibromialgíaGuadalupe ToskiNessuna valutazione finora
- Neuropatías Miopatias MiasteniaDocumento12 pagineNeuropatías Miopatias MiasteniaBianca BittiNessuna valutazione finora
- Patrones de HerenciaDocumento58 paginePatrones de HerenciaPaco RomeroNessuna valutazione finora
- Medicina ocupacional y rehabilitación en parálisis cerebral infantilDocumento49 pagineMedicina ocupacional y rehabilitación en parálisis cerebral infantilPaola Jhazmin Rivas GuerreroNessuna valutazione finora
- Enfermedades NeuromuscularesDocumento48 pagineEnfermedades NeuromuscularesJuan Martin CadelagoNessuna valutazione finora
- Niño HipotónicoDocumento44 pagineNiño HipotónicoEliza Magaly Contreras SamaniegoNessuna valutazione finora
- Clase 7 - Parálisis Cerebral InfantilDocumento38 pagineClase 7 - Parálisis Cerebral InfantilRosa Villegas Tello100% (1)
- Semana 12 Equinoterapia 2023-IDocumento37 pagineSemana 12 Equinoterapia 2023-ILeydi PaccoriNessuna valutazione finora
- Capitulo 112,113,114 RoccaDocumento16 pagineCapitulo 112,113,114 RoccaDaniela SinisterraNessuna valutazione finora
- Posito 2Documento2 paginePosito 2Denishitop Lopez JimenezNessuna valutazione finora
- Posito 2Documento2 paginePosito 2Denishitop Lopez JimenezNessuna valutazione finora
- MANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIDocumento70 pagineMANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIMac Perez Lopez100% (1)
- MANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIDocumento70 pagineMANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIMac Perez Lopez100% (1)
- Programa de Estudios de Tecnología Médica: Silabo Internado en Terapia Física Y RehabilitaciónDocumento10 paginePrograma de Estudios de Tecnología Médica: Silabo Internado en Terapia Física Y RehabilitaciónDenishitop Lopez JimenezNessuna valutazione finora
- Barreras Geograficas en La Salud y Poblacion PeruanaDocumento16 pagineBarreras Geograficas en La Salud y Poblacion PeruanaDenishitop Lopez Jimenez50% (2)
- Planeacion Estrategica Power PointDocumento41 paginePlaneacion Estrategica Power PointGladiz Yaneth CalitoNessuna valutazione finora
- Sistema Limbico y Autonomo OriginalDocumento53 pagineSistema Limbico y Autonomo OriginalDenishitop Lopez JimenezNessuna valutazione finora
- 17 Genu Varo y Genu ValgoDocumento2 pagine17 Genu Varo y Genu ValgoRomina Veronica AraozNessuna valutazione finora
- (2015) .D.informe de Laboratorio Febrero y Marzo-FebDocumento5 pagine(2015) .D.informe de Laboratorio Febrero y Marzo-FebDenishitop Lopez JimenezNessuna valutazione finora
- Ortesis y Ayudas para La MarchaDocumento72 pagineOrtesis y Ayudas para La MarchaJuan Rivera Wharton100% (2)
- Componente Salud - Guías para La RBC OMS - 2012Documento22 pagineComponente Salud - Guías para La RBC OMS - 2012Denishitop Lopez JimenezNessuna valutazione finora
- 2010 - E Tores-FISIOTERAPIA RESPIRATORIA EN NEONATOS PDFDocumento5 pagine2010 - E Tores-FISIOTERAPIA RESPIRATORIA EN NEONATOS PDFNydia Stirling100% (1)
- Abordaje Fisioterapeutico en Lesionados MedularesDocumento50 pagineAbordaje Fisioterapeutico en Lesionados MedularesDenishitop Lopez JimenezNessuna valutazione finora
- Procedimiento Atencion Radiodiagnostico PDFDocumento8 pagineProcedimiento Atencion Radiodiagnostico PDFDenishitop Lopez JimenezNessuna valutazione finora
- Accidente Cerebro VascularDocumento50 pagineAccidente Cerebro VascularDenishitop Lopez JimenezNessuna valutazione finora
- MANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIDocumento70 pagineMANUAL PARA LA VIGILANCIA DEL DESARROLLO INFANTIL (0-6 AñOS) EN EL CONTEXTO DE AIEPIMac Perez Lopez100% (1)
- Area Evolutivas Trabajoa AvanceDocumento8 pagineArea Evolutivas Trabajoa AvanceDenishitop Lopez JimenezNessuna valutazione finora
- Barreras Geograficas en La Salud y Poblacion PeruanaDocumento16 pagineBarreras Geograficas en La Salud y Poblacion PeruanaDenishitop Lopez Jimenez50% (2)
- Procedimiento Atencion Radiodiagnostico PDFDocumento8 pagineProcedimiento Atencion Radiodiagnostico PDFDenishitop Lopez JimenezNessuna valutazione finora
- Trabajito de Kan 2222Documento14 pagineTrabajito de Kan 2222Denishitop Lopez JimenezNessuna valutazione finora
- Barreras Geograficas en La Salud y Poblacion PeruanaDocumento16 pagineBarreras Geograficas en La Salud y Poblacion PeruanaDenishitop Lopez Jimenez50% (2)
- Abordaje Fisioterapeutico en Lesionados MedularesDocumento50 pagineAbordaje Fisioterapeutico en Lesionados MedularesDenishitop Lopez JimenezNessuna valutazione finora
- Accidente Cerebro VascularDocumento50 pagineAccidente Cerebro VascularDenishitop Lopez JimenezNessuna valutazione finora
- Abordaje Fisioteràpeutico en ADocumento68 pagineAbordaje Fisioteràpeutico en ADenishitop Lopez Jimenez100% (1)
- A 0Documento9 pagineA 0Denishitop Lopez JimenezNessuna valutazione finora
- Modelo PoiDocumento94 pagineModelo PoiMichell AmpueroNessuna valutazione finora
- Fnu U2 Atr KatmDocumento5 pagineFnu U2 Atr KatmKaty tm100% (2)
- Discapacidad: InstructivoDocumento30 pagineDiscapacidad: InstructivoGisele LuengasNessuna valutazione finora
- Prevencion de AdiccionesDocumento46 paginePrevencion de AdiccionesbarbatusjungNessuna valutazione finora
- ReportDocumento1 paginaReportWiliam Ernesto Vega FariasNessuna valutazione finora
- Cápsula Reflexiva #104Documento2 pagineCápsula Reflexiva #104Ludwing33Nessuna valutazione finora
- NOM 015 para La Prevención, Tratamiento y Control de La Diabetes Mellitus.Documento46 pagineNOM 015 para La Prevención, Tratamiento y Control de La Diabetes Mellitus.Pakofranco Ironhide PerceptorNessuna valutazione finora
- Caso Clinico Labio LeporinoDocumento4 pagineCaso Clinico Labio LeporinoMichel Angélica Betancur Figueroa0% (1)
- Trastorno Negativista DesafianteDocumento13 pagineTrastorno Negativista DesafianteDanny CalderonNessuna valutazione finora
- Pae FX Te Tibia y PeroneDocumento26 paginePae FX Te Tibia y PeroneJazmine Fasanando RamirezNessuna valutazione finora
- Enfermedades EruptivasDocumento3 pagineEnfermedades EruptivasGonza SpolidoriNessuna valutazione finora
- Oncologia Basica 2011 - SyllabusDocumento7 pagineOncologia Basica 2011 - SyllabusmrfnixeNessuna valutazione finora
- Apendicitis AgudaDocumento57 pagineApendicitis Agudatomshamilon21Nessuna valutazione finora
- Sepsis Neonatal: Causas, Diagnóstico y TratamientoDocumento36 pagineSepsis Neonatal: Causas, Diagnóstico y TratamientoBierce SáNessuna valutazione finora
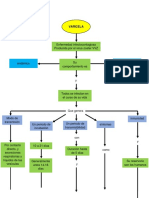
- Mapa Conseptual VaricelaDocumento2 pagineMapa Conseptual Varicelajhon daza mojicaNessuna valutazione finora
- Perfil HepaticoDocumento22 paginePerfil HepaticoRichard David Aguirre Barrientos100% (2)
- GPC 548 Asma Infantil Osteba Compl CaducDocumento162 pagineGPC 548 Asma Infantil Osteba Compl CaducTatty GrzNessuna valutazione finora
- Capítulo 1-Introduccion PDFDocumento8 pagineCapítulo 1-Introduccion PDFMike FontaneNessuna valutazione finora
- Infografia Mitos y Verdades de La Leche Materna OctubreDocumento3 pagineInfografia Mitos y Verdades de La Leche Materna OctubreMARTHA ROCIO MORALES100% (2)
- Hemorragias primera mitad embarazoDocumento6 pagineHemorragias primera mitad embarazoLiliana PinosNessuna valutazione finora
- Viernes, 2 de Octubre de 2020 5:05 P. MDocumento13 pagineViernes, 2 de Octubre de 2020 5:05 P. MCamilaNessuna valutazione finora
- Tema 1 Concepto y Alcance de La DietoterapiaDocumento8 pagineTema 1 Concepto y Alcance de La DietoterapiaYodyNessuna valutazione finora
- Amigdalitis AgudaDocumento19 pagineAmigdalitis AgudaFlor GonzalesNessuna valutazione finora
- Content ServerDocumento12 pagineContent ServerLuis Ángel ApancoNessuna valutazione finora
- Historia de La MedicinaDocumento12 pagineHistoria de La MedicinaRaymond TorresNessuna valutazione finora
- Norma de Diagnostico y Tratamiento de OftalmologiaDocumento169 pagineNorma de Diagnostico y Tratamiento de OftalmologiafelipeNessuna valutazione finora
- Clausulado Poliza Integral Estudiantil.Documento41 pagineClausulado Poliza Integral Estudiantil.Leandro TovarNessuna valutazione finora
- GlaucomaDocumento49 pagineGlaucomaRenato José CasartelliNessuna valutazione finora
- Actualización en DiabetesDocumento40 pagineActualización en DiabetesMarianella HerreraNessuna valutazione finora