Potrebbero piacerti anche
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5795)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Grain & Feed Milling Technology - November - December FULL EDITIONDocumento78 pagineGrain & Feed Milling Technology - November - December FULL EDITIONMilling and Grain magazineNessuna valutazione finora
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- A Hebrew Primer BallDocumento288 pagineA Hebrew Primer Ballondoveo4749Nessuna valutazione finora
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- S&R PreviewDocumento13 pagineS&R PreviewTiggasNessuna valutazione finora
- Eizenberg S General Anatomy PDFDocumento255 pagineEizenberg S General Anatomy PDFNikita Leanne FernandesNessuna valutazione finora
- Under The Sparkling SeaDocumento48 pagineUnder The Sparkling SeaCreepygamer this is power100% (3)
- Reliability Centered MaintenanceDocumento16 pagineReliability Centered MaintenanceDaniel OmolewaNessuna valutazione finora
- Stages of Evolution of ManDocumento3 pagineStages of Evolution of ManTristan Nialler100% (1)
- RADA in Business - Effective Communication ManualDocumento37 pagineRADA in Business - Effective Communication ManualGabriel Mendoza VargasNessuna valutazione finora
- Standard TestDocumento3 pagineStandard TestRhys JansenNessuna valutazione finora
- Che326 11 12ADocumento155 pagineChe326 11 12ADaniel OmolewaNessuna valutazione finora
- Lecture Notes On Petrchemicals From Refining and Cracked Petroleum ResiduesDocumento9 pagineLecture Notes On Petrchemicals From Refining and Cracked Petroleum ResiduesDaniel OmolewaNessuna valutazione finora
- Hazard Identification & Risk AssessmentDocumento19 pagineHazard Identification & Risk AssessmentDaniel OmolewaNessuna valutazione finora
- Manufacturing of Pharmceuticals: Dr. Ojewumi M.EDocumento8 pagineManufacturing of Pharmceuticals: Dr. Ojewumi M.EDaniel OmolewaNessuna valutazione finora
- Che 524-Module 1Documento44 pagineChe 524-Module 1Daniel OmolewaNessuna valutazione finora
- Enzyme Inhibition and ToxicityDocumento12 pagineEnzyme Inhibition and ToxicityDaniel OmolewaNessuna valutazione finora
- Kinetics of Microbial Growth: Dr. Ojewumi M.EDocumento15 pagineKinetics of Microbial Growth: Dr. Ojewumi M.EDaniel OmolewaNessuna valutazione finora
- Che 516 Rate-Controlling StepDocumento35 pagineChe 516 Rate-Controlling StepDaniel OmolewaNessuna valutazione finora
- Chm121 Module 1Documento21 pagineChm121 Module 1Daniel OmolewaNessuna valutazione finora
- SWEP 2016 Lecture NotesDocumento10 pagineSWEP 2016 Lecture NotesDaniel Omolewa100% (1)
- Hypothesis TestingDocumento7 pagineHypothesis TestingDaniel OmolewaNessuna valutazione finora
- Solution To Assignment 1Documento3 pagineSolution To Assignment 1Daniel Omolewa50% (2)
- IJEACS Paper4 - CorrectionsDocumento8 pagineIJEACS Paper4 - CorrectionsDaniel OmolewaNessuna valutazione finora
- Interpretation of Results: Ao Fa Fa+Fb - A FB Fa+Fb - BDocumento3 pagineInterpretation of Results: Ao Fa Fa+Fb - A FB Fa+Fb - BDaniel OmolewaNessuna valutazione finora
- 400l Alpha Semester Exams TimetableDocumento1 pagina400l Alpha Semester Exams TimetableDaniel OmolewaNessuna valutazione finora
- CHE 325 (Module 3)Documento53 pagineCHE 325 (Module 3)Daniel OmolewaNessuna valutazione finora
- WILD GIRLS: A Novel by Mary Stewart Atwell (Excerpt)Documento25 pagineWILD GIRLS: A Novel by Mary Stewart Atwell (Excerpt)Simon and Schuster50% (2)
- Earth and Life Sciences SLHTQ2 3Documento9 pagineEarth and Life Sciences SLHTQ2 3Melissa GerzonNessuna valutazione finora
- Plaza Mascotas-Lista de Precios PDFDocumento32 paginePlaza Mascotas-Lista de Precios PDFCarolina SaninNessuna valutazione finora
- Dark Hunters PDFDocumento5 pagineDark Hunters PDFMatthew HaydenNessuna valutazione finora
- Urinary System-1Documento48 pagineUrinary System-1Muhammad SajjadNessuna valutazione finora
- Business PlanDocumento11 pagineBusiness PlanArshit Mahajan100% (1)
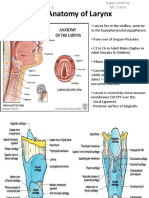
- Anatomy of Larynx - Wai Sheng Xuan Batch 3 1101A11688Documento27 pagineAnatomy of Larynx - Wai Sheng Xuan Batch 3 1101A11688Roshan Gill100% (1)
- The Mice That Set The Elephants FreeDocumento2 pagineThe Mice That Set The Elephants FreePeter LuaNessuna valutazione finora
- Effects of Feeding Dietary Palm Kernel Cake On,,pptxDocumento17 pagineEffects of Feeding Dietary Palm Kernel Cake On,,pptxAhmad BasriNessuna valutazione finora
- Rembrandt Corpus BueyDocumento24 pagineRembrandt Corpus BueyManuel100% (1)
- Customer Satisfaction of DairyDocumento118 pagineCustomer Satisfaction of DairynitindeoraNessuna valutazione finora
- Ihn General Training Reading Practice Test 6 Section 1 Questions 1 - 13Documento14 pagineIhn General Training Reading Practice Test 6 Section 1 Questions 1 - 13Anonymous f5qGAcZYNessuna valutazione finora
- General Awareness and Aptitude Test: Questions: 40 Marks: 160Documento58 pagineGeneral Awareness and Aptitude Test: Questions: 40 Marks: 160Nooruddin Sheik100% (2)
- Pemeriksaan Anemia SMP Darul MaarifDocumento38 paginePemeriksaan Anemia SMP Darul MaarifPuskesmas BanyuputihNessuna valutazione finora
- Shri Ganesh Chaturthi - 29.08.2014, Friday, Hasta Star, Kanya RasiDocumento16 pagineShri Ganesh Chaturthi - 29.08.2014, Friday, Hasta Star, Kanya RasiastrokpmNessuna valutazione finora
- Alphabet Adventures - Lyrics To The SongsDocumento7 pagineAlphabet Adventures - Lyrics To The SongsmaryleesunseriNessuna valutazione finora
- (1951) Out of Step: Events in The Two Lives of An Anti-Jewish Camel-DoctorDocumento71 pagine(1951) Out of Step: Events in The Two Lives of An Anti-Jewish Camel-DoctorHerbert Hillary Booker 2ndNessuna valutazione finora
- Chimp Haven Federal Complaint 6-13-22Documento11 pagineChimp Haven Federal Complaint 6-13-22USA TODAY NetworkNessuna valutazione finora
- DAT Bootcamp - Developmental BiologyDocumento6 pagineDAT Bootcamp - Developmental BiologyPearl PascuaNessuna valutazione finora
- Forensic Chemistry Lecture #11Documento25 pagineForensic Chemistry Lecture #11Michael QuidorNessuna valutazione finora
- Eng-B w10 InternetDocumento6 pagineEng-B w10 InternetmoralassNessuna valutazione finora
- Cicipu Dictionary PDFDocumento188 pagineCicipu Dictionary PDFFrancisco José Da SilvaNessuna valutazione finora