Potrebbero piacerti anche
- Level 3 Cert PT Course Manual 2019-UnlockedDocumento130 pagineLevel 3 Cert PT Course Manual 2019-UnlockedNIKOS MOUMOURIS100% (2)
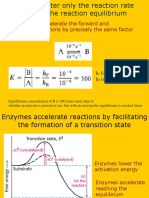
- Enzyme Kinetics - InhibitionDocumento40 pagineEnzyme Kinetics - InhibitionRodney Baldwin100% (1)
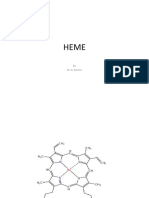
- Heme Metabolism PDFDocumento19 pagineHeme Metabolism PDFAnonymous jW7BU44ACNessuna valutazione finora
- Carbohydrates NotesDocumento70 pagineCarbohydrates NotesRavindra AgarwalNessuna valutazione finora
- Amino Acid Metabolism II 10-15-08Documento35 pagineAmino Acid Metabolism II 10-15-08Timothy George100% (1)
- BIO307 Lecture 5 (Enzyme Kinetics I)Documento11 pagineBIO307 Lecture 5 (Enzyme Kinetics I)Phenyo Mmereki100% (1)
- Infection and Infectious ProcessDocumento44 pagineInfection and Infectious Processpandey omkarNessuna valutazione finora
- Koss y Honda - Citología Del Tracto Urinario (2012)Documento142 pagineKoss y Honda - Citología Del Tracto Urinario (2012)Lord Verminaard Luis Carreño DuranNessuna valutazione finora
- NBME 15 QuizletDocumento12 pagineNBME 15 Quizletrmelendez00192% (12)
- Enzyme KineticsDocumento26 pagineEnzyme KineticsLyra LasangreNessuna valutazione finora
- Microbiology NotesDocumento103 pagineMicrobiology NotesNetskie Batac100% (1)
- ProteinDocumento39 pagineProteinNICHOLE MOJELLO100% (2)
- Edexcel Ial Biology Lab BookpdfDocumento69 pagineEdexcel Ial Biology Lab Bookpdfbgtes12367% (6)
- The Structure and Function of MacromoleculesDocumento70 pagineThe Structure and Function of MacromoleculesLustried Nadyang100% (1)
- Answers Chapter 10 and 11 ExamDocumento7 pagineAnswers Chapter 10 and 11 ExamMara Romanoff75% (4)
- Design Arguments For The Existence of GodDocumento19 pagineDesign Arguments For The Existence of GodCollins Rotich100% (2)
- Biochemistry Saeed Oraby Part 3Documento276 pagineBiochemistry Saeed Oraby Part 3أسہہآمہهہ شہعہلآن100% (3)
- CH11-Amino Acid MetabolismDocumento106 pagineCH11-Amino Acid MetabolismChatchawinNessuna valutazione finora
- INMO Air User GuideDocumento34 pagineINMO Air User GuidebagasNessuna valutazione finora
- Identification of Business Processes in An Enterprise ManagementDocumento10 pagineIdentification of Business Processes in An Enterprise ManagementWilliam GunawanNessuna valutazione finora
- Quipper 1 - Earth and Life Science - CellDocumento17 pagineQuipper 1 - Earth and Life Science - CellPhil Christian MerinoNessuna valutazione finora
- Labster MidtermsDocumento4 pagineLabster MidtermsMaria Sophia Faith100% (1)
- EnzymesDocumento6 pagineEnzymesBenedique Valdez0% (1)
- Oxidative PhosphorylationDocumento33 pagineOxidative PhosphorylationJithendra Babu0% (1)
- Nzymes: By: Mrs. Kalaivani Sathish. M. Pharm, Assistant Professor, Pims - PanipatDocumento63 pagineNzymes: By: Mrs. Kalaivani Sathish. M. Pharm, Assistant Professor, Pims - Panipaturmila pandeyNessuna valutazione finora
- Types of FermentersDocumento3 pagineTypes of FermentersHoney krishnaNessuna valutazione finora
- Metabolism of DisaccharidesDocumento15 pagineMetabolism of DisaccharidesminaNessuna valutazione finora
- Reducing and Non-Reducing SugarDocumento4 pagineReducing and Non-Reducing SugarAyunee ZulhasNessuna valutazione finora
- GluconeogenesisDocumento11 pagineGluconeogenesisMithilesh RautNessuna valutazione finora
- Lipid MetabolismDocumento78 pagineLipid MetabolismGhaidaa SadeqNessuna valutazione finora
- Microbial NutritionDocumento8 pagineMicrobial NutritionManohar PattarNessuna valutazione finora
- Metabolism of HemeDocumento138 pagineMetabolism of HemeZafir MonNessuna valutazione finora
- Enzyme SpecificityDocumento6 pagineEnzyme SpecificityfayeNessuna valutazione finora
- Carbohydrate Chemistry: Dr. Herat D. Soni Assistant Professor Rural Medical College LoniDocumento117 pagineCarbohydrate Chemistry: Dr. Herat D. Soni Assistant Professor Rural Medical College LoniWwwanand111Nessuna valutazione finora
- Digestive System Gizi Andreanyta MelialaDocumento105 pagineDigestive System Gizi Andreanyta Melialaannaafia69969Nessuna valutazione finora
- Osmosis in The Digestive SystemDocumento1 paginaOsmosis in The Digestive SystemNadhiraAzhar_Nessuna valutazione finora
- Antibiotic Resistance in Bacteria 1Documento32 pagineAntibiotic Resistance in Bacteria 1Sandeep MandalNessuna valutazione finora
- Heme SynthasisDocumento47 pagineHeme SynthasisAmjad AlmousawiNessuna valutazione finora
- Biochem 9Documento4 pagineBiochem 9Abdullah RaufNessuna valutazione finora
- Heme Metabolism HarperDocumento14 pagineHeme Metabolism HarperHOD gtmcNessuna valutazione finora
- Ch21 Conversion of Amino Acids To Specialized ProductsDocumento62 pagineCh21 Conversion of Amino Acids To Specialized Productsendale gebregzabherNessuna valutazione finora
- PorphyrinsDocumento14 paginePorphyrinsVytheeshwaran Vedagiri100% (2)
- Heme SynthesisDocumento51 pagineHeme SynthesisGanesh HansdahNessuna valutazione finora
- Biosynthesis of HemoglobinDocumento42 pagineBiosynthesis of Hemoglobin95kodok85Nessuna valutazione finora
- Biomolecules (Introduction, Structure and Functions) : Smita Rastogi & U. N. DwivediDocumento17 pagineBiomolecules (Introduction, Structure and Functions) : Smita Rastogi & U. N. DwivediEmmanuel UgwuNessuna valutazione finora
- BCH 301 - Porphyrins and PorphyriasDocumento12 pagineBCH 301 - Porphyrins and Porphyriasoseghalemercy409Nessuna valutazione finora
- Topic 12. Porphirin Metabolism.Documento7 pagineTopic 12. Porphirin Metabolism.Manar BehiNessuna valutazione finora
- Porphyrin Metbolism 2023Documento55 paginePorphyrin Metbolism 2023kenaabebe87Nessuna valutazione finora
- Or Deamination PLP Also Participates in Reactions in Which An Amino Group IsDocumento5 pagineOr Deamination PLP Also Participates in Reactions in Which An Amino Group IsNissa zuzulNessuna valutazione finora
- Heme Synthesis: DR Amina Tariq BiochemistryDocumento21 pagineHeme Synthesis: DR Amina Tariq BiochemistryMadeline UdarbeNessuna valutazione finora
- Functions of GlycogenDocumento11 pagineFunctions of GlycogenBea SamonteNessuna valutazione finora
- By Dr. A. KwenaDocumento67 pagineBy Dr. A. KwenaJacob MasikaNessuna valutazione finora
- Molecular Genetics and Metabolism: John D. PhillipsDocumento14 pagineMolecular Genetics and Metabolism: John D. PhillipsJaSNessuna valutazione finora
- B.Pharm, 5 Semester Pharmacognosy and Phytochemistry-II (Theory) Subject Code: BP 504 T Unit-IDocumento18 pagineB.Pharm, 5 Semester Pharmacognosy and Phytochemistry-II (Theory) Subject Code: BP 504 T Unit-IFahim UddinNessuna valutazione finora
- Heme SynthesisDocumento32 pagineHeme SynthesisHari PrasathNessuna valutazione finora
- University of Gujrat (UOG) 06-04-2012Documento17 pagineUniversity of Gujrat (UOG) 06-04-2012Sachin SharmaNessuna valutazione finora
- 2.2. Biosintesis Hemoglobin PDFDocumento48 pagine2.2. Biosintesis Hemoglobin PDFIkaTriRahayuNessuna valutazione finora
- Protein Turnover and Amino Acid Catabolism: Chem 454: Biochemistry II University of Wisconsin-Eau ClaireDocumento54 pagineProtein Turnover and Amino Acid Catabolism: Chem 454: Biochemistry II University of Wisconsin-Eau ClaireArif Mc TomsNessuna valutazione finora
- NOTES Tutorial 4 Proteins RevisionNotes BMSC2120Documento5 pagineNOTES Tutorial 4 Proteins RevisionNotes BMSC2120ZAFFAR QURESHINessuna valutazione finora
- Porphyrias: M. F. M. James and R. J. HiftDocumento11 paginePorphyrias: M. F. M. James and R. J. HiftJhonnathan RodriguezNessuna valutazione finora
- Lec 2.5 Dr. Ngadikun 2016 PorfirinDocumento33 pagineLec 2.5 Dr. Ngadikun 2016 PorfirinRobertOktaChandraNessuna valutazione finora
- Subjective QuestionsDocumento5 pagineSubjective QuestionsAgustina MandasariNessuna valutazione finora
- BiochemistryDocumento2 pagineBiochemistryJessyca MarvellaNessuna valutazione finora
- Amino Acid MetabolismDocumento15 pagineAmino Acid Metabolismshanto.tn98Nessuna valutazione finora
- Acetate, Mevalonate and Shikimic Acid PathwaysDocumento19 pagineAcetate, Mevalonate and Shikimic Acid Pathwaysamitaggarwal7888% (8)
- Topic: Structure and Function of Haemoglobin: GC University, LahoreDocumento8 pagineTopic: Structure and Function of Haemoglobin: GC University, LahoreAbdullah MunawarNessuna valutazione finora
- Biosintesis Asam Amino & Nukleotida: Puji Lestari Prodi Kimia FST UNSOEDDocumento35 pagineBiosintesis Asam Amino & Nukleotida: Puji Lestari Prodi Kimia FST UNSOEDJennie JisooNessuna valutazione finora
- METABOLISME ASAM AMINO Protein BiologiDocumento31 pagineMETABOLISME ASAM AMINO Protein BiologiAxzchiuu :vNessuna valutazione finora
- Folate and Cobalamin MetabolismDocumento5 pagineFolate and Cobalamin MetabolismheyahroxNessuna valutazione finora
- Plant Cell - WikipediaDocumento32 paginePlant Cell - WikipediaSherelyn Bernardo100% (1)
- Chapter 2 CK-12 Biology Chapter 2 Worksheets PDFDocumento23 pagineChapter 2 CK-12 Biology Chapter 2 Worksheets PDFIlincaVasilescuNessuna valutazione finora
- Assignment No. 01: Human Digestive System and Respiratory SystemDocumento3 pagineAssignment No. 01: Human Digestive System and Respiratory SystemZIA UR REHMANNessuna valutazione finora
- What Professor Armand Marie Leroi Doesn't KnowDocumento1 paginaWhat Professor Armand Marie Leroi Doesn't Know'Messianic' DylanologistNessuna valutazione finora
- Materi Reading Ke 3Documento5 pagineMateri Reading Ke 3viara nindaNessuna valutazione finora
- Taksonomi NumerikDocumento17 pagineTaksonomi NumerikMohammad Toni MardiansyahNessuna valutazione finora
- Holy Family Nursing BSC HonsDocumento126 pagineHoly Family Nursing BSC HonsPiyush Singh RajputNessuna valutazione finora
- MCQsDocumento32 pagineMCQsamNessuna valutazione finora
- Cleavage: Cleavage Is A Series of Extremely Rapid Mitotic Divisions That Immediately Follow FertilizationDocumento14 pagineCleavage: Cleavage Is A Series of Extremely Rapid Mitotic Divisions That Immediately Follow Fertilizationkashif manzoorNessuna valutazione finora
- Science 9 W1-W4Documento21 pagineScience 9 W1-W4Jennifer Lacadue Masoa - LumantasNessuna valutazione finora
- Origin of MetazoaDocumento4 pagineOrigin of MetazoaAmod KumarNessuna valutazione finora
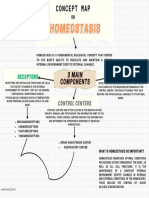
- Concept Map Group 2Documento1 paginaConcept Map Group 2mike jesterNessuna valutazione finora
- Reviewer in Advance Biology 4th QuarterDocumento6 pagineReviewer in Advance Biology 4th QuarterTherese CuetoNessuna valutazione finora
- (9783110411324 - Biochemistry Laboratory Manual For Undergraduates) Biochemistry Laboratory Manual For Undergraduates PDFDocumento186 pagine(9783110411324 - Biochemistry Laboratory Manual For Undergraduates) Biochemistry Laboratory Manual For Undergraduates PDFBritt NeyNessuna valutazione finora
- MarcotDocumento2 pagineMarcotZonalyne Tangbawaniddoba PascuaDelacruzNessuna valutazione finora
- Biology OPDocumento1.478 pagineBiology OPSasha RasinNessuna valutazione finora
- Science10 Q4 Week-3-BiomoleculesDocumento7 pagineScience10 Q4 Week-3-BiomoleculesMaskter ArcheryNessuna valutazione finora
- 10 week:: Archaeal and Eukaryotic Molecular Biology (와 진핵생물의 분자생물학)Documento26 pagine10 week:: Archaeal and Eukaryotic Molecular Biology (와 진핵생물의 분자생물학)MoonHoLeeNessuna valutazione finora
- Diabetic NephropathyDocumento41 pagineDiabetic NephropathyBen IntosiusNessuna valutazione finora
- Human CD Chart PDFDocumento1 paginaHuman CD Chart PDFLia WieNessuna valutazione finora
- MBoC5e - CHPT - 13 Intracellular Vesicular TrafficDocumento111 pagineMBoC5e - CHPT - 13 Intracellular Vesicular TrafficAnnisaazharNessuna valutazione finora