Potrebbero piacerti anche
- Introduction to Machine Learning in the Cloud with Python: Concepts and PracticesDa EverandIntroduction to Machine Learning in the Cloud with Python: Concepts and PracticesNessuna valutazione finora
- Support Vector Machine: Fundamentals and ApplicationsDa EverandSupport Vector Machine: Fundamentals and ApplicationsNessuna valutazione finora
- Time Series Analysis and Its Applications: With R Examples: Second EditionDocumento18 pagineTime Series Analysis and Its Applications: With R Examples: Second EditionPaula VilaNessuna valutazione finora
- Stata Graph Library For Network AnalysisDocumento41 pagineStata Graph Library For Network AnalysisAlexander SainesNessuna valutazione finora
- Forecast Time Series With R LanguageDocumento98 pagineForecast Time Series With R Languageec04017Nessuna valutazione finora
- Using The R Commander A Point-And-Click Interface For The R by Fox, JohnDocumento238 pagineUsing The R Commander A Point-And-Click Interface For The R by Fox, JohnAlex GHNessuna valutazione finora
- Seefeld-Statistics Using R With Biological Examples PDFDocumento325 pagineSeefeld-Statistics Using R With Biological Examples PDFManuel LoGaNessuna valutazione finora
- Outliers in Time Series DataDocumento8 pagineOutliers in Time Series DataJalluNessuna valutazione finora
- Dendrogramas Con RDocumento24 pagineDendrogramas Con RWR SalasNessuna valutazione finora
- 4-PCA and SVDDocumento32 pagine4-PCA and SVDHiteshNessuna valutazione finora
- Handling and Processing Strings in R PDFDocumento113 pagineHandling and Processing Strings in R PDFInstantRamenNessuna valutazione finora
- Functions in R Sem-III 2021 PDFDocumento30 pagineFunctions in R Sem-III 2021 PDFrajveer shahNessuna valutazione finora
- OrdinalexampleR PDFDocumento9 pagineOrdinalexampleR PDFDamon CopelandNessuna valutazione finora
- CleaningData Chapter 1 With RDocumento21 pagineCleaningData Chapter 1 With RMahmoud TriguiNessuna valutazione finora
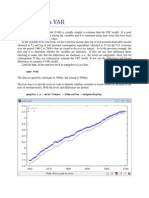
- Estimating A VAR - GretlDocumento9 pagineEstimating A VAR - Gretlkaddour7108Nessuna valutazione finora
- Data Science With RDocumento21 pagineData Science With RErick Dimas Cirilo BerazainNessuna valutazione finora
- Arch Model and Time-Varying VolatilityDocumento17 pagineArch Model and Time-Varying VolatilityJorge Vega RodríguezNessuna valutazione finora
- Notes For Multivariate Statistics With RDocumento189 pagineNotes For Multivariate Statistics With RAhmed AssalNessuna valutazione finora
- Introduction To Ggplot2: Saier (Vivien) Ye September 16, 2013Documento32 pagineIntroduction To Ggplot2: Saier (Vivien) Ye September 16, 201310yangb92Nessuna valutazione finora
- Data Science Course ContentDocumento8 pagineData Science Course ContentQshore online trainingNessuna valutazione finora
- Five College RDocumento104 pagineFive College Ret_phonehome_2Nessuna valutazione finora
- Czekanowski Index-Based Similarity As Alternative Correlation Measure in N-Asset Portfolio AnalysisDocumento1 paginaCzekanowski Index-Based Similarity As Alternative Correlation Measure in N-Asset Portfolio AnalysisKonrad KonefałNessuna valutazione finora
- R Reference Card: 1 Getting Started 3 Input and OutputDocumento7 pagineR Reference Card: 1 Getting Started 3 Input and OutputanamartinfernandezNessuna valutazione finora
- Dplyr TutorialDocumento22 pagineDplyr TutorialDamini KapoorNessuna valutazione finora
- Creating A Live World Weather Map Using Shiny - by M. Makkawi - The Startup - MediumDocumento40 pagineCreating A Live World Weather Map Using Shiny - by M. Makkawi - The Startup - MediumDirga DanielNessuna valutazione finora
- R Data Analyst DARDocumento6 pagineR Data Analyst DARvaishu-Nessuna valutazione finora
- R For Health Data ScienceDocumento365 pagineR For Health Data ScienceragmarNessuna valutazione finora
- Exploratory Data AnalysisDocumento38 pagineExploratory Data Analysishss601Nessuna valutazione finora
- Use R For Climate ResearchDocumento31 pagineUse R For Climate ResearchJames WallaceNessuna valutazione finora
- Sas AnalyticsDocumento35 pagineSas AnalyticsprakashnethaNessuna valutazione finora
- R For Programmers PDFDocumento370 pagineR For Programmers PDFfaibistesNessuna valutazione finora
- A Little Book of R For Time SeriesDocumento75 pagineA Little Book of R For Time Series정관용Nessuna valutazione finora
- Practical Missing Data Analysis in SPSSDocumento19 paginePractical Missing Data Analysis in SPSSlphouneNessuna valutazione finora
- GSEMModellingusingStata PDFDocumento97 pagineGSEMModellingusingStata PDFmarikum74Nessuna valutazione finora
- Matrix Decomposition and Its Application in Statistics NKDocumento82 pagineMatrix Decomposition and Its Application in Statistics NKDaniel KozakevichNessuna valutazione finora
- Image Analysis AlgorithmDocumento27 pagineImage Analysis AlgorithmEngr EbiNessuna valutazione finora
- Non Linear RegressionDocumento20 pagineNon Linear RegressionPrasad TpNessuna valutazione finora
- Exponential DistributionDocumento19 pagineExponential DistributionArabi Ali ANessuna valutazione finora
- Handbook Bio StatDocumento305 pagineHandbook Bio StatADWINDT100% (1)
- Ap, GP, HPDocumento29 pagineAp, GP, HPJay DayalNessuna valutazione finora
- R Handout Statistics and Data Analysis Using RDocumento91 pagineR Handout Statistics and Data Analysis Using Rtele6Nessuna valutazione finora
- Time Series PennDocumento67 pagineTime Series PennVishnu Prakash SinghNessuna valutazione finora
- History of ProbabilityDocumento17 pagineHistory of ProbabilityonlypingNessuna valutazione finora
- Decision Trees For Predictive Modeling (Neville)Documento24 pagineDecision Trees For Predictive Modeling (Neville)Mohith Reddy100% (1)
- CopulaDocumento21 pagineCopulahghaedi66Nessuna valutazione finora
- Lean Pub Data Wrangling With RDocumento245 pagineLean Pub Data Wrangling With RHector Perez VilcapazaNessuna valutazione finora
- Learning Ggplot2 PDFDocumento39 pagineLearning Ggplot2 PDFMaria NovosolovNessuna valutazione finora
- 03 A Logistic Regression 2018Documento13 pagine03 A Logistic Regression 2018Reham TarekNessuna valutazione finora
- Introduction To Data Mining With Case Studies - Sample IndexDocumento16 pagineIntroduction To Data Mining With Case Studies - Sample IndexJosé A. Ferreira Queimada0% (1)
- 1.4 Muon PhysicDocumento19 pagine1.4 Muon PhysicExfalNessuna valutazione finora
- Using MS Project 2010Documento222 pagineUsing MS Project 2010Georgi StoyanovNessuna valutazione finora
- Goals, Process, and Challenges of Exploratory Data Analysis: An Interview StudyDocumento10 pagineGoals, Process, and Challenges of Exploratory Data Analysis: An Interview StudyRACHNA JAINNessuna valutazione finora
- Learning Statistics With RDocumento564 pagineLearning Statistics With RDanielNessuna valutazione finora
- Longitudinal Research: Present Status and Future Prospects: John B. Willett & Judith D. SingerDocumento19 pagineLongitudinal Research: Present Status and Future Prospects: John B. Willett & Judith D. SingerTeflon SlimNessuna valutazione finora
- Pub Applied Statistics Principles and ExamplesDocumento196 paginePub Applied Statistics Principles and Examplesraghudv100% (1)
- Computational Statistics With RDocumento125 pagineComputational Statistics With Rkjhjhlkhlkjtiuytututh100% (1)
- 45 Genetic AlgorithmsDocumento20 pagine45 Genetic AlgorithmsJayeng WidiatmokoNessuna valutazione finora
- The Diagnostic Process: Graphic Approach to Probability and Inference in Clinical MedicineDa EverandThe Diagnostic Process: Graphic Approach to Probability and Inference in Clinical MedicineNessuna valutazione finora
- Start Predicting In A World Of Data Science And Predictive AnalysisDa EverandStart Predicting In A World Of Data Science And Predictive AnalysisNessuna valutazione finora
- Agresti Ordinal TutorialDocumento75 pagineAgresti Ordinal TutorialKen MatsudaNessuna valutazione finora
- II &IV Semester PG Notification-2020Documento3 pagineII &IV Semester PG Notification-2020soma1243Nessuna valutazione finora
- Probationary Engineer (Optics) - 2016Documento4 pagineProbationary Engineer (Optics) - 2016huzaifa17iNessuna valutazione finora
- Performance Appraisal at WiproDocumento16 paginePerformance Appraisal at WiproPratik D. Patel100% (1)
- 01 Amaravati Logo CompetitionDocumento4 pagine01 Amaravati Logo Competitionsoma1243Nessuna valutazione finora
- NASA Labs FacilitiesDocumento27 pagineNASA Labs Facilitiessoma1243Nessuna valutazione finora
- Sri Venkateswara University:: Tirupati: 07.01.2019 ThursdayDocumento1 paginaSri Venkateswara University:: Tirupati: 07.01.2019 Thursdaysoma1243Nessuna valutazione finora
- Gorbachev Hadoop RDocumento55 pagineGorbachev Hadoop Rsoma1243Nessuna valutazione finora
- SIZE YoYDocumento4 pagineSIZE YoYsoma1243Nessuna valutazione finora
- Report Entry SchemaDocumento3 pagineReport Entry Schemasoma1243Nessuna valutazione finora
- Iata CodesDocumento860 pagineIata Codessoma1243Nessuna valutazione finora
- SIZE QoQDocumento4 pagineSIZE QoQsoma1243Nessuna valutazione finora
- Destin Code ExecDocumento2 pagineDestin Code Execsoma1243Nessuna valutazione finora
- WS Insert QueryDocumento47 pagineWS Insert Querysoma1243Nessuna valutazione finora
- Index FormulaDocumento1 paginaIndex Formulasoma1243Nessuna valutazione finora
- Academics 140713032558 Phpapp01Documento82 pagineAcademics 140713032558 Phpapp01soma1243Nessuna valutazione finora
- Destin Code ExecDocumento2 pagineDestin Code Execsoma1243Nessuna valutazione finora
- Project 120519125806 Phpapp01Documento52 pagineProject 120519125806 Phpapp01ranasiddharthNessuna valutazione finora
- Class-1 Assignment-1 Calculate The Student Grades For MR Thomas - 2Documento1 paginaClass-1 Assignment-1 Calculate The Student Grades For MR Thomas - 2soma1243Nessuna valutazione finora
- Notification SSCMPR Technical Asst Superintendent Other PostsDocumento30 pagineNotification SSCMPR Technical Asst Superintendent Other Postsumesh72Nessuna valutazione finora
- 13 ZIJMR Vol2 Issue5 May 2012Documento11 pagine13 ZIJMR Vol2 Issue5 May 2012Dipak ThakurNessuna valutazione finora
- Edureka Presentation Java Essentials For Hadoop Class 3Documento53 pagineEdureka Presentation Java Essentials For Hadoop Class 3soma1243Nessuna valutazione finora
- Microsoft SQL Server 2012 Installation GuideDocumento31 pagineMicrosoft SQL Server 2012 Installation GuideArdit MeziniNessuna valutazione finora
- Tcs Company DetailsDocumento1 paginaTcs Company DetailscanapathyNessuna valutazione finora
- Os Log Process Hadoop PDFDocumento8 pagineOs Log Process Hadoop PDFsoma1243Nessuna valutazione finora
- Cloudera Connector For TableauDocumento12 pagineCloudera Connector For Tableausoma1243Nessuna valutazione finora
- MBA II Semester NotificationDocumento2 pagineMBA II Semester Notificationsoma1243Nessuna valutazione finora
- Cloudera ODBC Driver For Apache Hive Install Guide 2-5-13Documento58 pagineCloudera ODBC Driver For Apache Hive Install Guide 2-5-13soma1243Nessuna valutazione finora
- Hive For SQL Users: Cheat SheetDocumento3 pagineHive For SQL Users: Cheat Sheetsrikanth07balusuNessuna valutazione finora
- MBA II Semester NotificationDocumento2 pagineMBA II Semester Notificationsoma1243Nessuna valutazione finora
- Gender and Informal Sector Employment in Indonesia-Hlm313-316Documento10 pagineGender and Informal Sector Employment in Indonesia-Hlm313-316suyanto sandiatmoNessuna valutazione finora
- 1 s2.0 S2666154322001958 MainDocumento8 pagine1 s2.0 S2666154322001958 MainAndrés GutiérrezNessuna valutazione finora
- The Modification of Jury Instructions To Improve Juror Verdicts and Confession Recognitions in A Criminal Trial by Meghan PasalaDocumento9 pagineThe Modification of Jury Instructions To Improve Juror Verdicts and Confession Recognitions in A Criminal Trial by Meghan PasalaNahid HossainNessuna valutazione finora
- Predictive Modelling Project Report FinalDocumento49 paginePredictive Modelling Project Report FinalNandakumar33% (9)
- Applied Business Analytics: Starts JUNE 30, 2021 3 Months - OnlineDocumento11 pagineApplied Business Analytics: Starts JUNE 30, 2021 3 Months - OnlinekrishnaNessuna valutazione finora
- Zavgren 1985 PDFDocumento27 pagineZavgren 1985 PDFdr musafirNessuna valutazione finora
- Tax Audit'effectivenessDocumento14 pagineTax Audit'effectivenessRaga VenthiniNessuna valutazione finora
- Metabolic Syndrome and Associated Factors in Older People: A Secondary Analysis of SABE ColombiaDocumento16 pagineMetabolic Syndrome and Associated Factors in Older People: A Secondary Analysis of SABE ColombiaLuis Carlos Venegas SanabriaNessuna valutazione finora
- Migrasi Ulang Alik PDFDocumento7 pagineMigrasi Ulang Alik PDFBudi Susetyo HutomoNessuna valutazione finora
- Measuring Eating Competence: Psychometric Properties and Validity of The Ecsatter InventoryDocumento13 pagineMeasuring Eating Competence: Psychometric Properties and Validity of The Ecsatter InventoryMacarena VargasNessuna valutazione finora
- Financial Literacy and Financial Well Being Among Generation Z University Students Evidence From GreeceDocumento23 pagineFinancial Literacy and Financial Well Being Among Generation Z University Students Evidence From GreeceTiffany Joy MoloNessuna valutazione finora
- Case QuestionsDocumento2 pagineCase Questionssan75% (4)
- Champions of Change and Strategic Shifts: The Role of Internal and External Change AdvocatesDocumento19 pagineChampions of Change and Strategic Shifts: The Role of Internal and External Change AdvocatesAlex RiveraNessuna valutazione finora
- Review On Credit Card Fraud Detection Using Machine Learning AlgorithmsDocumento5 pagineReview On Credit Card Fraud Detection Using Machine Learning AlgorithmsGiuseppeLegrottaglieNessuna valutazione finora
- BinningDocumento14 pagineBinningSher Win100% (1)
- Results Binomial Logistic Regression: Model Deviance AIC RDocumento7 pagineResults Binomial Logistic Regression: Model Deviance AIC RSmit MangeNessuna valutazione finora
- Department of Economics: ECONOMICS 481: Economics Research Paper and SeminarDocumento15 pagineDepartment of Economics: ECONOMICS 481: Economics Research Paper and SeminarHan ZhongNessuna valutazione finora
- Isivi PDFDocumento86 pagineIsivi PDFanilsidhNessuna valutazione finora
- Healthcare 10 01438 v3Documento14 pagineHealthcare 10 01438 v3Zeera MohamadNessuna valutazione finora
- Study of Mobility Impact Through Cable Car Implementation in Bandung, IndonesiaDocumento121 pagineStudy of Mobility Impact Through Cable Car Implementation in Bandung, IndonesiaSheryta ArsalliaNessuna valutazione finora
- Credit Card Fraudulent Transaction Detection and PreventionDocumento8 pagineCredit Card Fraudulent Transaction Detection and PreventionIJRASETPublications100% (1)
- 1 s2.0 S1042443123000690 MainDocumento17 pagine1 s2.0 S1042443123000690 MainOumema AL AZHARNessuna valutazione finora
- A Review A Review of Financial Accounting Fraud Detection Based On Data Mining Techniquesof Financial Accounting Fraud Detection Based On Data Mining TechniquesDocumento11 pagineA Review A Review of Financial Accounting Fraud Detection Based On Data Mining Techniquesof Financial Accounting Fraud Detection Based On Data Mining TechniqueskhuongcomputerNessuna valutazione finora
- Binomial Logistic Regression: Julia HartmanDocumento78 pagineBinomial Logistic Regression: Julia HartmanMonny ManolacheNessuna valutazione finora
- Bahan 3Documento25 pagineBahan 3Bagas Tri PangestuNessuna valutazione finora
- Using Mplus: In: Using Mplus For Structural Equation Modeling: A Researcher's GuideDocumento15 pagineUsing Mplus: In: Using Mplus For Structural Equation Modeling: A Researcher's Guidemesay83Nessuna valutazione finora
- 5700 MlogitDocumento38 pagine5700 MlogitNawaz AliNessuna valutazione finora
- Aden Idiris 2017 ResearchpaperDocumento26 pagineAden Idiris 2017 ResearchpaperyousafNessuna valutazione finora