Potrebbero piacerti anche
- The Shameless Fraud Known As Darwinian EvolutionDocumento37 pagineThe Shameless Fraud Known As Darwinian EvolutionJeremy James100% (2)
- FILE 20230308 111109 Tutor-Genetics-solutionDocumento25 pagineFILE 20230308 111109 Tutor-Genetics-solutionthuytrang21032004Nessuna valutazione finora
- The Hollow Earth-BernardDocumento95 pagineThe Hollow Earth-BernardAmadeo Rosi de los CamposNessuna valutazione finora
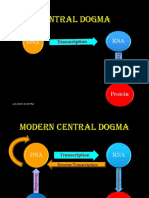
- Signature Assignment 4 Central Dogma 1Documento5 pagineSignature Assignment 4 Central Dogma 1api-709276885Nessuna valutazione finora
- Shahuindo Geologic Analysis Report FinalDocumento69 pagineShahuindo Geologic Analysis Report FinalWilly Lopez Mogrovejo0% (1)
- The Radix System: Vivian E. Robson, B.SCDocumento114 pagineThe Radix System: Vivian E. Robson, B.SCJack GannNessuna valutazione finora
- Tutorial Solution Chapter 1+2+3Documento28 pagineTutorial Solution Chapter 1+2+3Nguyen Minh HoangNessuna valutazione finora
- Pangaea Ultima Futuro E#8f8Documento95 paginePangaea Ultima Futuro E#8f8Luis GonçalvesNessuna valutazione finora
- Earth and Life ScienceDocumento4 pagineEarth and Life ScienceArmelin AlipayoNessuna valutazione finora
- Astrology and MarriageDocumento32 pagineAstrology and MarriageprngalaNessuna valutazione finora
- FLC2000 and Wellbore StrengtheningDocumento10 pagineFLC2000 and Wellbore StrengtheningKinni ShenoldNessuna valutazione finora
- Graha YudhaDocumento40 pagineGraha YudhaRamesh kumar100% (1)
- S11 12ES Ia e 1Documento6 pagineS11 12ES Ia e 1Erica De Guzman AngelesNessuna valutazione finora
- B.V. Gueorguiev Annotated Catalogue of The Carabid Beetles of Albania Coleoptera Carabidae v. 64 Pensoft Series Faunistica 2007Documento245 pagineB.V. Gueorguiev Annotated Catalogue of The Carabid Beetles of Albania Coleoptera Carabidae v. 64 Pensoft Series Faunistica 2007ismetjakuNessuna valutazione finora
- Genetic Coding PDFDocumento12 pagineGenetic Coding PDFMobileLegends SbNessuna valutazione finora
- Exercises Genetics USTH2022Documento15 pagineExercises Genetics USTH2022yungiang157Nessuna valutazione finora
- MutasiDocumento39 pagineMutasiY VismitanandaNessuna valutazione finora
- Aman NyeDocumento13 pagineAman NyeSolah AlaamNessuna valutazione finora
- An Introduction To GeneticsDocumento43 pagineAn Introduction To Geneticsrifatalib4u3504Nessuna valutazione finora
- Beta Globulin Gene SequenceDocumento5 pagineBeta Globulin Gene SequenceB Riaz AhmedNessuna valutazione finora
- Appendix 2 - The Genetic Code - 2018 - Human BiochemistryDocumento2 pagineAppendix 2 - The Genetic Code - 2018 - Human Biochemistrynour achkarNessuna valutazione finora
- Mutation & MutagenesisDocumento20 pagineMutation & MutagenesisMuhammad FarooqNessuna valutazione finora
- 08 Genes and MutationDocumento16 pagine08 Genes and Mutationapi-255000301Nessuna valutazione finora
- Statistics For Microarrays: Biological Background: Gene Expression and Molecular Laboratory TechniquesDocumento47 pagineStatistics For Microarrays: Biological Background: Gene Expression and Molecular Laboratory TechniquesVarun DwivediNessuna valutazione finora
- Central Dogma: DNA RNADocumento42 pagineCentral Dogma: DNA RNAGayu KjNessuna valutazione finora
- PM703 Practical Biotechnology (2019) PM703 Practical Biotechnology (2019)Documento20 paginePM703 Practical Biotechnology (2019) PM703 Practical Biotechnology (2019)Hadeer DaifAllahNessuna valutazione finora
- Bioinformatics Seminar3rdOct18Documento25 pagineBioinformatics Seminar3rdOct18subhasree majumderNessuna valutazione finora
- Transcription and TranslationDocumento53 pagineTranscription and TranslationIndranilNessuna valutazione finora
- Transferrin: Cdna Chromosomal Localization : Human Characterization andDocumento5 pagineTransferrin: Cdna Chromosomal Localization : Human Characterization andAndrimencitNessuna valutazione finora
- Gene ExpressionDocumento1 paginaGene ExpressionMigs BernalNessuna valutazione finora
- Practicals in GeneticsDocumento112 paginePracticals in GeneticscarlosaeserranoNessuna valutazione finora
- Maam Hindi Ko Po Alam Saan Kukunin To Kaya Di Ko Po Nilagyan NG Answer, Sorry Po.Documento2 pagineMaam Hindi Ko Po Alam Saan Kukunin To Kaya Di Ko Po Nilagyan NG Answer, Sorry Po.DONNA MAE LOBERESNessuna valutazione finora
- Protein English Aug 2006Documento13 pagineProtein English Aug 2006Rimal IsaacNessuna valutazione finora
- Lec No 4Documento18 pagineLec No 4Janet BarcimoNessuna valutazione finora
- Development of ARMS PCR Tests For Detection of ComDocumento7 pagineDevelopment of ARMS PCR Tests For Detection of ComKevin JebarajNessuna valutazione finora
- RevewipacketanswerkeyrevisedDocumento5 pagineRevewipacketanswerkeyrevisedapi-235160519Nessuna valutazione finora
- Gene MutationDocumento19 pagineGene Mutationمحمد رعدNessuna valutazione finora
- Gene, Proteins, and Genetic CodeDocumento37 pagineGene, Proteins, and Genetic CodejuniorNessuna valutazione finora
- CL662 PW 02 Gene FindingDocumento39 pagineCL662 PW 02 Gene FindingKanupriya TiwariNessuna valutazione finora
- pmTFP1 ClathrinDocumento4 paginepmTFP1 ClathrinAlleleBiotechNessuna valutazione finora
- Mutations WorksheetDocumento2 pagineMutations WorksheetSarah LowtherNessuna valutazione finora
- Omputational ENE Rediction: Cse/Bimm/Beng 181 M 24, 2011Documento45 pagineOmputational ENE Rediction: Cse/Bimm/Beng 181 M 24, 2011Raghav SureshNessuna valutazione finora
- Cloning Characterization Specific Androgen Receptor ProstateDocumento5 pagineCloning Characterization Specific Androgen Receptor ProstateMaria Luiza CraciunNessuna valutazione finora
- MBBS1 Workshop 2 2021 Sequential v2Documento64 pagineMBBS1 Workshop 2 2021 Sequential v2K ZarabianNessuna valutazione finora
- Mutation: - Today? Mutation Refers To Changes That Occur Within GenesDocumento18 pagineMutation: - Today? Mutation Refers To Changes That Occur Within Genesapplemango93Nessuna valutazione finora
- Escherichia Coli DH10BDocumento2 pagineEscherichia Coli DH10BSecc. 2 Marco Antonio Aviles RomeroNessuna valutazione finora
- DNA Sequencing: How Do You Do It?Documento11 pagineDNA Sequencing: How Do You Do It?IqbalFahmiJuwonoNessuna valutazione finora
- Protein English Aug 2006Documento18 pagineProtein English Aug 2006lordniklausNessuna valutazione finora
- CEFA8 D 01Documento56 pagineCEFA8 D 01idhem1110Nessuna valutazione finora
- Transcription Worksheet 2Documento2 pagineTranscription Worksheet 2John Philip NapalNessuna valutazione finora
- Apa Și Biologia MolecularăDocumento157 pagineApa Și Biologia MolecularăClaudia MateiNessuna valutazione finora
- Protein Microarrays: Michael SnyderDocumento44 pagineProtein Microarrays: Michael Snyderbb27Nessuna valutazione finora
- Mitochondrial Genome Testudinae 2Documento7 pagineMitochondrial Genome Testudinae 2JULIO CÉSAR CHÁVEZ GALARZANessuna valutazione finora
- Pharma Lec 2ADocumento22 paginePharma Lec 2AQusay Al MaghayerhNessuna valutazione finora
- Backcross N Genetic CodeDocumento6 pagineBackcross N Genetic Codehafizah_90Nessuna valutazione finora
- Continuity of Life & Genetic ControlDocumento25 pagineContinuity of Life & Genetic ControlYudi Santoso0% (1)
- Bioinformatics LAb ReportDocumento7 pagineBioinformatics LAb ReportBriana Halbert100% (3)
- Artículo 1Documento10 pagineArtículo 1AoriwishNessuna valutazione finora
- QGenetics Tutorial 7Documento1 paginaQGenetics Tutorial 7wend23Nessuna valutazione finora
- Ing Gen P3Documento11 pagineIng Gen P3Bruno Geanpiero Díaz CruzNessuna valutazione finora
- pmTFP1 ZyxinDocumento5 paginepmTFP1 ZyxinAlleleBiotechNessuna valutazione finora
- Molecular Mechanism of MutationDocumento55 pagineMolecular Mechanism of MutationDiotima BhattacharyaNessuna valutazione finora
- Genetic Code HMDocumento30 pagineGenetic Code HMdrhmpatel100% (2)
- Student Name: Lesson 7: DNA and Genes DataDocumento3 pagineStudent Name: Lesson 7: DNA and Genes DataMinh Nguyen DucNessuna valutazione finora
- Virtual Lab-Point MutationsDocumento3 pagineVirtual Lab-Point MutationsAndy Rodriguez0% (1)
- A Quantitative PCR Method For Measuring Absolute Telomere LengthDocumento10 pagineA Quantitative PCR Method For Measuring Absolute Telomere LengthKaiswan GanNessuna valutazione finora
- Xpressed Equence Ag: Ests - OutlineDocumento26 pagineXpressed Equence Ag: Ests - OutlineSatyam SinghNessuna valutazione finora
- Ion Channel Factsbook: Extracellular Ligand-Gated ChannelsDa EverandIon Channel Factsbook: Extracellular Ligand-Gated ChannelsNessuna valutazione finora
- Fast Facts: Les troubles d'oxydation des acides gras à chaîne longue: Comprendre, identifier et aiderDa EverandFast Facts: Les troubles d'oxydation des acides gras à chaîne longue: Comprendre, identifier et aiderNessuna valutazione finora
- Methods For Preclinical Evaluation of Bioactive Natural ProductsDa EverandMethods For Preclinical Evaluation of Bioactive Natural ProductsNessuna valutazione finora
- Electrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsDa EverandElectrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsNessuna valutazione finora
- PHD Ordinance 2013Documento20 paginePHD Ordinance 2013Yougesh KumarNessuna valutazione finora
- MSC MPlil ZoologyDocumento18 pagineMSC MPlil ZoologyYougesh KumarNessuna valutazione finora
- WWW - Ccsuniversity.ac - in New Common-Syllabus ZoologyDocumento20 pagineWWW - Ccsuniversity.ac - in New Common-Syllabus ZoologyYougesh KumarNessuna valutazione finora
- Executive Summary Major Research ProjectDocumento13 pagineExecutive Summary Major Research ProjectYougesh KumarNessuna valutazione finora
- Price List 2010Documento120 paginePrice List 2010Yougesh KumarNessuna valutazione finora
- GreatpyramidjeezaDocumento608 pagineGreatpyramidjeezapierce033033Nessuna valutazione finora
- Foreland PHD ThesisDocumento4 pagineForeland PHD ThesisSEDRAZNADNessuna valutazione finora
- Deterministic Chaos in General Relativity PDFDocumento472 pagineDeterministic Chaos in General Relativity PDFPriti Gupta100% (1)
- Radioactive Decay WorksheetDocumento4 pagineRadioactive Decay WorksheetSuta PinatihNessuna valutazione finora
- Lunar Eclipses 2001-2100Documento5 pagineLunar Eclipses 2001-2100sidhanti26Nessuna valutazione finora
- PGTL Pendahuluan 2018Documento36 paginePGTL Pendahuluan 2018Nurselina SimarmataNessuna valutazione finora
- Flood Rou1Documento13 pagineFlood Rou1rockstaraliNessuna valutazione finora
- Grade 11 & 12 2nd Prelim ExamDocumento2 pagineGrade 11 & 12 2nd Prelim ExamHealth NutritionNessuna valutazione finora
- Biology BeedDocumento2 pagineBiology BeedRj Nerf Monteverde CalasangNessuna valutazione finora
- DYING ON ROMAN ROADS - Inter Vias Latron PDFDocumento18 pagineDYING ON ROMAN ROADS - Inter Vias Latron PDFKaterinaLogotheti100% (1)
- Experiment 4Documento7 pagineExperiment 4Sunil FranklinNessuna valutazione finora
- 2014 Sanjuan Chinchon Cemento-PortlandDocumento182 pagine2014 Sanjuan Chinchon Cemento-PortlandJon Peres PeresNessuna valutazione finora
- Sedimentary RockDocumento65 pagineSedimentary RockAbdul Moeed KalsonNessuna valutazione finora
- Prepared By: Orlan Defensor BalanoDocumento59 paginePrepared By: Orlan Defensor BalanoOrlan Defensor BalanoNessuna valutazione finora
- DSC New Rules 2012Documento20 pagineDSC New Rules 2012navn7650% (2)
- GR 9 Natural Science ANS SHEET Eng PDFDocumento8 pagineGR 9 Natural Science ANS SHEET Eng PDFJillrey RegondolaNessuna valutazione finora
- Anchoring EncyclopediaDocumento78 pagineAnchoring EncyclopediaHamidullah Asady WardakNessuna valutazione finora
- Tolleranze IsoDocumento6 pagineTolleranze IsoAngela RossoniNessuna valutazione finora