Potrebbero piacerti anche
- Sickle Cell AnemiaDocumento37 pagineSickle Cell Anemiahazelposis75% (4)
- Aplastic Anemia Pa Tho PhysiologyDocumento3 pagineAplastic Anemia Pa Tho Physiologyneil052275% (4)
- Iron Deficiency Anemia PathoDocumento6 pagineIron Deficiency Anemia PathoENessuna valutazione finora
- AnemiaDocumento32 pagineAnemiaSherina Christo100% (2)
- Idiopathic Thrombocytopenic Purpura Case StudyDocumento38 pagineIdiopathic Thrombocytopenic Purpura Case Studygiadda100% (9)
- Anemia PathophysiologyDocumento2 pagineAnemia PathophysiologyHoney Lorie D. Simbajon67% (6)
- Pathophysiology Sickle Cell AnemiaDocumento1 paginaPathophysiology Sickle Cell AnemiaTine GuibaoNessuna valutazione finora
- HELLP Concept Map RevisedDocumento1 paginaHELLP Concept Map RevisedwandaNessuna valutazione finora
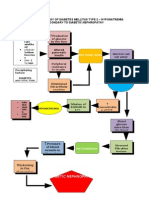
- Pathophysiology of Diabetes Mellitus Type 2Documento1 paginaPathophysiology of Diabetes Mellitus Type 2faula rocamora100% (3)
- Pathophysiology Case 4 Sickle Cell AnemiaDocumento4 paginePathophysiology Case 4 Sickle Cell AnemiaKARL MARLU LUZANessuna valutazione finora
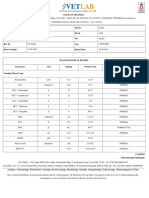
- CBC InterpretationDocumento6 pagineCBC InterpretationKate Basa100% (1)
- PathophysiologyDocumento4 paginePathophysiologyAngelou Joefred Congreso100% (1)
- Aplastic Anemia Lecture 1aDocumento39 pagineAplastic Anemia Lecture 1aniaaseta100% (2)
- Nephrotic Case Study With AnswersDocumento3 pagineNephrotic Case Study With AnswersKBNessuna valutazione finora
- Hematology FinalDocumento103 pagineHematology Finalallanrnmanaloto100% (1)
- Sickle Cell AnemiaDocumento63 pagineSickle Cell Anemiaoss-20502745100% (4)
- Sickle Cell AnemiaDocumento52 pagineSickle Cell AnemiahudaNessuna valutazione finora
- CellulitisDocumento12 pagineCellulitisAlma Bertos-Agub100% (1)
- Summary of All AnemiaDocumento2 pagineSummary of All Anemiabenlarsena93% (14)
- Malaria Case StudyDocumento6 pagineMalaria Case StudymichpaduaNessuna valutazione finora
- Electrolyte Imbalance 1Documento3 pagineElectrolyte Imbalance 1Marius Clifford BilledoNessuna valutazione finora
- Pathophysiology Sickle Cell Anemia PDFDocumento1 paginaPathophysiology Sickle Cell Anemia PDFTine GuibaoNessuna valutazione finora
- Novilyn C. Pataray BSN - Ii Hemophilia: St. Paul College of Ilocos SurDocumento1 paginaNovilyn C. Pataray BSN - Ii Hemophilia: St. Paul College of Ilocos SurCharina AubreyNessuna valutazione finora
- Sicle Cell Concept MapDocumento1 paginaSicle Cell Concept MapRosa100% (1)
- CKD PathophysiologyDocumento1 paginaCKD Pathophysiologylloyd_santino67% (3)
- Chronic Renal FailureDocumento37 pagineChronic Renal Failuredorkiebaby100% (10)
- Leukemia)Documento66 pagineLeukemia)Arianne BugnaNessuna valutazione finora
- Pathophysiology of Acute Myelogenous Leukemia Fab m4Documento3 paginePathophysiology of Acute Myelogenous Leukemia Fab m4KristaMaeC.Lazo100% (2)
- Pathophysiology of Aplastic AnemiaDocumento1 paginaPathophysiology of Aplastic Anemiatinatin989100% (2)
- Acute Glomerulonephritis Case StudyDocumento12 pagineAcute Glomerulonephritis Case StudyPrincess Tindugan100% (1)
- Concept Map On CVA Bleed Left Thalamo-Ganglionic Bleed Patient Name: Mr. DGDDocumento1 paginaConcept Map On CVA Bleed Left Thalamo-Ganglionic Bleed Patient Name: Mr. DGDBert Brian Bolido100% (1)
- Megaloblastic Anemia: A Case StudyDocumento30 pagineMegaloblastic Anemia: A Case Studyromeo rivera100% (3)
- Nephrotic SyndromeDocumento36 pagineNephrotic SyndromedrtpkNessuna valutazione finora
- Reason For Needing Health Care: Key Problem / ND: Noncompliance Key Problem / NDDocumento6 pagineReason For Needing Health Care: Key Problem / ND: Noncompliance Key Problem / NDnursing concept mapsNessuna valutazione finora
- Pathophysiology in Liver CirrhosisDocumento4 paginePathophysiology in Liver CirrhosisCyrus Ortalla RobinNessuna valutazione finora
- Schistosomiasis Case StudyDocumento5 pagineSchistosomiasis Case Studyapi-318749549Nessuna valutazione finora
- Community Acquired Pneumonia: Ang Gobonseng, Ed Gerard R. MedclerkDocumento66 pagineCommunity Acquired Pneumonia: Ang Gobonseng, Ed Gerard R. MedclerkRyn Anfone100% (2)
- MeaslesDocumento88 pagineMeasleserilarchi100% (1)
- Acute Lymphocytic LeukemiaDocumento12 pagineAcute Lymphocytic Leukemiajustin_saneNessuna valutazione finora
- CBC Interpetation Dr. MuslehDocumento37 pagineCBC Interpetation Dr. MuslehMusleh Al MusalhiNessuna valutazione finora
- Clinical Manifestations and Treatment of HypokalemiaDocumento16 pagineClinical Manifestations and Treatment of Hypokalemiagerontogeria100% (2)
- Hemophilia PathophysiologyDocumento79 pagineHemophilia Pathophysiologyjeba100% (8)
- Pathophysiology of Liver CirrhosisDocumento2 paginePathophysiology of Liver Cirrhosisgaelty100% (4)
- Pathophysiology Dengue 2Documento4 paginePathophysiology Dengue 2KatherineNessuna valutazione finora
- PathoDocumento7 paginePathoAnonymous 87fNoO2fhVNessuna valutazione finora
- Thalassemia Case ReportDocumento40 pagineThalassemia Case ReportGracelia DamanikNessuna valutazione finora
- Pathophysiology HypokalemiaDocumento1 paginaPathophysiology HypokalemiaRnspeakcomNessuna valutazione finora
- Non-Modifiable Risk Factors: Modifiable Risk FactorsDocumento6 pagineNon-Modifiable Risk Factors: Modifiable Risk FactorsNeil Andro Marcelo100% (1)
- Electrolytes & FluidimbalancesDocumento80 pagineElectrolytes & FluidimbalancesDennis Nyambane MomanyiNessuna valutazione finora
- Leukemia With PathophysiologyDocumento34 pagineLeukemia With Pathophysiologymabec pagaduan90% (41)
- Pathophysiology of Benign Prostatic HyperplasiaDocumento1 paginaPathophysiology of Benign Prostatic HyperplasiaKevin Jade Herrera0% (2)
- Hematology: FANER, Ned Denebe LACANILAO, Sunshine NUCUM, Billie Kim PAGADUAN, Maribec PUA, MonalisaDocumento31 pagineHematology: FANER, Ned Denebe LACANILAO, Sunshine NUCUM, Billie Kim PAGADUAN, Maribec PUA, MonalisatzuquinoNessuna valutazione finora
- Hematology Part 1 NotesDocumento23 pagineHematology Part 1 NotesDr. Benson BenjaminNessuna valutazione finora
- P.07 ANEMIA Dr. Buliyat 09 03 18Documento5 pagineP.07 ANEMIA Dr. Buliyat 09 03 18Raymund Dan AldabaNessuna valutazione finora
- AnemiaDocumento7 pagineAnemiaoktavianprNessuna valutazione finora
- Microcytic Type Aeitology Clinical Features Investigations ManagementDocumento7 pagineMicrocytic Type Aeitology Clinical Features Investigations ManagementJason AnthonyNessuna valutazione finora
- Blood Pharmacology PDFDocumento13 pagineBlood Pharmacology PDFManikanta GupthaNessuna valutazione finora
- Hemapathology Case: Adrian Joe G. CaballesDocumento15 pagineHemapathology Case: Adrian Joe G. CaballesAdrian CaballesNessuna valutazione finora
- Blood FunctionsDocumento3 pagineBlood FunctionshelloaNessuna valutazione finora
- III. FINAL Nursing Care of Clients With Disturbances in Male Female Reproduction Sexuality EditedVersionDocumento26 pagineIII. FINAL Nursing Care of Clients With Disturbances in Male Female Reproduction Sexuality EditedVersionKurt PepitoNessuna valutazione finora
- Lymphomas With PathophysiologyDocumento30 pagineLymphomas With Pathophysiologymabec pagaduan91% (11)
- ARDS With PathophysiologyDocumento79 pagineARDS With Pathophysiologymabec pagaduan95% (19)
- Leukemia With PathophysiologyDocumento34 pagineLeukemia With Pathophysiologymabec pagaduan90% (41)
- Disseminated Intravascular Coagulation With PathophysiologyDocumento18 pagineDisseminated Intravascular Coagulation With Pathophysiologymabec pagaduan100% (6)
- Multiple Myeloma With PathophysiologyDocumento32 pagineMultiple Myeloma With Pathophysiologymabec pagaduan90% (10)
- Bio Ass I QPDocumento7 pagineBio Ass I QPSwayam GosaviNessuna valutazione finora
- Yoga Basics For Men PDFDocumento47 pagineYoga Basics For Men PDFfludor100% (1)
- Contact Point ContoursDocumento69 pagineContact Point ContourstarekrabiNessuna valutazione finora
- Studi Kasus: Pemalsuan Daging Sapi Dengan Daging Babi Hutan Di Kota Bogor Lailatun Nida, Herwin Pisestyani, Chaerul BasriDocumento10 pagineStudi Kasus: Pemalsuan Daging Sapi Dengan Daging Babi Hutan Di Kota Bogor Lailatun Nida, Herwin Pisestyani, Chaerul Basriclarentina aristawatiNessuna valutazione finora
- Trabecular Hepatocellular Carcinoma in A DogDocumento2 pagineTrabecular Hepatocellular Carcinoma in A Dogpradeep. mampilliNessuna valutazione finora
- Choanal Atresia PDFDocumento8 pagineChoanal Atresia PDFMonna Medani LysabellaNessuna valutazione finora
- Nursing Care Process Assessment Nursing Diagnosis ObjectiveDocumento6 pagineNursing Care Process Assessment Nursing Diagnosis ObjectiveKMNessuna valutazione finora
- Top Homeopathic Remedies For ColdsDocumento2 pagineTop Homeopathic Remedies For ColdsAkash kumarNessuna valutazione finora
- Modern Dental Assisting 11th Edition Bird Test BankDocumento16 pagineModern Dental Assisting 11th Edition Bird Test Bankhebexuyenod8q100% (31)
- Possessive AdjectivesDocumento15 paginePossessive AdjectivesMohammed ElouahalNessuna valutazione finora
- DUTY SDH + CKDDocumento6 pagineDUTY SDH + CKDadelia putri wirandaniNessuna valutazione finora
- Para Sample QuestionsDocumento5 paginePara Sample QuestionsMaria Christina LagartejaNessuna valutazione finora
- Test Bank For Ekg Plain and Simple 3rd Edition EllisDocumento8 pagineTest Bank For Ekg Plain and Simple 3rd Edition Elliscanebrutalfniy66Nessuna valutazione finora
- HIV-Associated Opportunistic Infections of The CNSDocumento13 pagineHIV-Associated Opportunistic Infections of The CNSmauroignacioNessuna valutazione finora
- Haematology, Biochemistry, Serology and Immunology, Microbiology, Cytology, Histopathology, Endocrinology, Ultrasonography & X-RayDocumento2 pagineHaematology, Biochemistry, Serology and Immunology, Microbiology, Cytology, Histopathology, Endocrinology, Ultrasonography & X-RayPankaj BeniwalNessuna valutazione finora
- Vitamins and Minerals: Nutrient (Vitamins) Needed For Key SourcesDocumento4 pagineVitamins and Minerals: Nutrient (Vitamins) Needed For Key SourcesKevin Carl A. CorpuzNessuna valutazione finora
- Why Is Rigor Mortis Absent in AnthraxDocumento25 pagineWhy Is Rigor Mortis Absent in Anthraxravigg100% (2)
- Checklist (PapSmear)Documento5 pagineChecklist (PapSmear)Sinung BawonoNessuna valutazione finora
- MP - Hardgainer GuideDocumento14 pagineMP - Hardgainer GuideAnthony Dinicolantonio100% (2)
- Occlusal AnalysisDocumento14 pagineOcclusal AnalysisDavid DongNessuna valutazione finora
- Upper Molar Distalization: A Critical Analysis: MF Sfondrini V Cacciafesta G SfondriniDocumento13 pagineUpper Molar Distalization: A Critical Analysis: MF Sfondrini V Cacciafesta G SfondriniCypry SchmitzNessuna valutazione finora
- Allergy Diagnostic TestingDocumento149 pagineAllergy Diagnostic TestingArrow Ou100% (1)
- Adrenal Tumor-Pheochromocytoma 10-10-19Documento30 pagineAdrenal Tumor-Pheochromocytoma 10-10-19JessicaNessuna valutazione finora
- Parasitology Case Presentation: Group 3Documento42 pagineParasitology Case Presentation: Group 3Christine Joy LegaspiNessuna valutazione finora
- CABERGOLINE Safety2Documento6 pagineCABERGOLINE Safety2Kane SmithNessuna valutazione finora
- Safety Tracker in Excel For HSE ProfessionalsDocumento22 pagineSafety Tracker in Excel For HSE Professionalsngomsia parfaitNessuna valutazione finora
- Prescription Pattern of Antibiotics Used in The Management of Respiratory Tract Infections in Paediatric PopulationDocumento5 paginePrescription Pattern of Antibiotics Used in The Management of Respiratory Tract Infections in Paediatric PopulationBaru Chandrasekhar RaoNessuna valutazione finora
- SEM2 MALARIA Plasmodium Falciparum 2013Documento23 pagineSEM2 MALARIA Plasmodium Falciparum 2013odysseyfairy2739Nessuna valutazione finora
- Sensations and Sensory Pathways General Senses Test Procedure Normal Result Abnormal Result Clinical InterpretationDocumento5 pagineSensations and Sensory Pathways General Senses Test Procedure Normal Result Abnormal Result Clinical InterpretationAbby MataNessuna valutazione finora
- Year 6 VocabularyDocumento7 pagineYear 6 VocabularyBala MuraliNessuna valutazione finora