Potrebbero piacerti anche
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Int Endodontic J - 2022 - Terauchi - Present Status and Future Directions Removal of Fractured InstrumentsDocumento25 pagineInt Endodontic J - 2022 - Terauchi - Present Status and Future Directions Removal of Fractured Instruments吳國豪Nessuna valutazione finora
- Space Time Continuum - Terence McKennaDocumento6 pagineSpace Time Continuum - Terence McKennaKali23YugaNessuna valutazione finora
- Engine Control SystemDocumento7 pagineEngine Control SystemFaisal Al HusainanNessuna valutazione finora
- Advanced Communication LaboratoryDocumento5 pagineAdvanced Communication LaboratoryJose DahlsonNessuna valutazione finora
- 2nd PUC Question Papers Physics 2006-2010Documento21 pagine2nd PUC Question Papers Physics 2006-2010Mohan Kumar P100% (1)
- Qualitative Analysis of CationsDocumento12 pagineQualitative Analysis of CationsRegina Morales0% (1)
- Tank Design CalculationDocumento20 pagineTank Design CalculationHairil HerliansyahNessuna valutazione finora
- Kim Lighting B30 Series Bollard Brochure 1985Documento20 pagineKim Lighting B30 Series Bollard Brochure 1985Alan MastersNessuna valutazione finora
- WINSEM2019-20 MEE1002 TH VL2019205001913 DA-1 QP KEY Assignment IDocumento9 pagineWINSEM2019-20 MEE1002 TH VL2019205001913 DA-1 QP KEY Assignment IDebdoot GhoshNessuna valutazione finora
- False-Position Method of Solving A Nonlinear Equation: Exact RootDocumento6 pagineFalse-Position Method of Solving A Nonlinear Equation: Exact Rootmacynthia26Nessuna valutazione finora
- Characteristics and Firing Control of Thyristor Controlled Series Compensation InstallationsDocumento5 pagineCharacteristics and Firing Control of Thyristor Controlled Series Compensation Installationsjm.mankavil6230Nessuna valutazione finora
- ECON1203 PASS Week 3Documento4 pagineECON1203 PASS Week 3mothermonkNessuna valutazione finora
- CV 101Documento4 pagineCV 101frco1504Nessuna valutazione finora
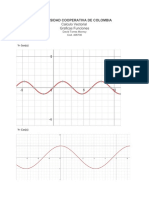
- Universidad Cooperativa de Colombia: Calculo Vectorial Graficas FuncionesDocumento11 pagineUniversidad Cooperativa de Colombia: Calculo Vectorial Graficas FuncionesDavid TorresNessuna valutazione finora
- MATH22558 Final Project - Winter 2018Documento4 pagineMATH22558 Final Project - Winter 2018HardilazizNessuna valutazione finora
- NA Curve FittingDocumento31 pagineNA Curve FittingRadwan HammadNessuna valutazione finora
- Lateral-Torsional Buckling: KiepahdusDocumento120 pagineLateral-Torsional Buckling: KiepahdusOrhan YanyatmazNessuna valutazione finora
- CNTH Skema Fizik Paper 3Documento3 pagineCNTH Skema Fizik Paper 3Norfadila Mat JusofNessuna valutazione finora
- 3 IntroductionDocumento5 pagine3 IntroductionKhamvanh PhengnaoneNessuna valutazione finora
- Umass Lowell Computer Science 91.503: Graduate AlgorithmsDocumento46 pagineUmass Lowell Computer Science 91.503: Graduate AlgorithmsShivam AtriNessuna valutazione finora
- My IT 2Documento56 pagineMy IT 2sentaljohnNessuna valutazione finora
- Sieve Analysis of Soil: By: Muhammad Firdaus, ST, MTDocumento12 pagineSieve Analysis of Soil: By: Muhammad Firdaus, ST, MTdaus_parisi100% (1)
- Assignment Submit by 1 May Humidification Operations: H A K ADocumento2 pagineAssignment Submit by 1 May Humidification Operations: H A K AHarshil ChangelaNessuna valutazione finora
- Engineering Syllabus of First Year EE, ECE, PWE, AEIE, EEE, IC, Apparel 2007Documento37 pagineEngineering Syllabus of First Year EE, ECE, PWE, AEIE, EEE, IC, Apparel 2007Anish LahiriNessuna valutazione finora
- An Elin Load Tap Changer Diagnosis by DgaDocumento4 pagineAn Elin Load Tap Changer Diagnosis by Dgamartinez_joselNessuna valutazione finora
- TB PhysicsTestY3 654b29bcc38e07.654b29bed4Documento17 pagineTB PhysicsTestY3 654b29bcc38e07.654b29bed4Prakash ManchukondaNessuna valutazione finora
- Bed Plate Main Engine BedplateDocumento52 pagineBed Plate Main Engine BedplateSuhas KassaNessuna valutazione finora
- Lab 2Documento5 pagineLab 2Adeem Hassan KhanNessuna valutazione finora
- Week11 Turton - Cost of ManufacturingDocumento32 pagineWeek11 Turton - Cost of ManufacturingNrl Nbl Rmln100% (1)
- Gold Nanoparticles ColorDocumento3 pagineGold Nanoparticles ColorBrandyNessuna valutazione finora