Potrebbero piacerti anche
- Analysis of Trend Following SystemsDocumento52 pagineAnalysis of Trend Following SystemsClement Li100% (1)
- Good Clinical PracticeDocumento129 pagineGood Clinical PracticeSreeraj Guruvayoor S100% (1)
- Principles and Practice of Clinical Trial MedicineDa EverandPrinciples and Practice of Clinical Trial MedicineValutazione: 4 su 5 stelle4/5 (1)
- Investigators Responsibilities With GCPDocumento16 pagineInvestigators Responsibilities With GCPLlosa JuneNessuna valutazione finora
- Handbook For GCPDocumento132 pagineHandbook For GCPanotherlevel88Nessuna valutazione finora
- The Sourcebook for Clinical Research: A Practical Guide for Study ConductDa EverandThe Sourcebook for Clinical Research: A Practical Guide for Study ConductValutazione: 5 su 5 stelle5/5 (1)
- Ethical Issues in Clinical ResearchDocumento33 pagineEthical Issues in Clinical ResearchSenthil ThyagarajanNessuna valutazione finora
- Equipment Qualification in the Pharmaceutical IndustryDa EverandEquipment Qualification in the Pharmaceutical IndustryValutazione: 3.5 su 5 stelle3.5/5 (3)
- Fundamentals of Biologicals Regulation: Vaccines and Biotechnology MedicinesDa EverandFundamentals of Biologicals Regulation: Vaccines and Biotechnology MedicinesValutazione: 5 su 5 stelle5/5 (1)
- Legal and Ethical Issues in ResearchDocumento5 pagineLegal and Ethical Issues in ResearchAditya SoniNessuna valutazione finora
- EthicsDocumento31 pagineEthicsALI KAMRANNessuna valutazione finora
- Accident Causation Theories and ConceptDocumento4 pagineAccident Causation Theories and ConceptShayne Aira AnggongNessuna valutazione finora
- Clinical Trial ProtocolDocumento30 pagineClinical Trial Protocolanand kumar gondNessuna valutazione finora
- By: Kris Traver and Nitin JainDocumento14 pagineBy: Kris Traver and Nitin JainMahesh BhagwatNessuna valutazione finora
- Research EthicsDocumento54 pagineResearch EthicsThiên Nhai Cô Khách67% (3)
- Research & Ethics CommitteeDocumento57 pagineResearch & Ethics CommitteeRajinderNessuna valutazione finora
- CVP Solution (Quiz)Documento9 pagineCVP Solution (Quiz)Angela Miles DizonNessuna valutazione finora
- Ethical Challenges of Research-Behavioral and Social Sciencies Research PDFDocumento66 pagineEthical Challenges of Research-Behavioral and Social Sciencies Research PDFcamelia_10i746Nessuna valutazione finora
- City Gas Distribution ReportDocumento22 pagineCity Gas Distribution Reportdimple1101100% (9)
- Ensuring National Biosecurity: Institutional Biosafety CommitteesDa EverandEnsuring National Biosecurity: Institutional Biosafety CommitteesCarole R BaskinNessuna valutazione finora
- Ethical Considerations When Preparing a Clinical Research ProtocolDa EverandEthical Considerations When Preparing a Clinical Research ProtocolNessuna valutazione finora
- Ethical Issues in Clinical ResearchDocumento23 pagineEthical Issues in Clinical ResearchDaxesh PatelNessuna valutazione finora
- Good Clinical PracticesDocumento75 pagineGood Clinical PracticesLuisaReyesNessuna valutazione finora
- Fundamentals and Applications of Renewable Energy by Mehmet Kanoglu, Yunus Cengel, John CimbalaDocumento413 pagineFundamentals and Applications of Renewable Energy by Mehmet Kanoglu, Yunus Cengel, John CimbalaFrancesco Nocera100% (1)
- International Regulatory Requirements On Clinical Trails and Data ManagementDocumento19 pagineInternational Regulatory Requirements On Clinical Trails and Data ManagementJay PraveenNessuna valutazione finora
- Clinical Research: Principles, Practice and PerspectiveDa EverandClinical Research: Principles, Practice and PerspectiveNessuna valutazione finora
- Introduction To Investigators Responsibilities With Good Clinical PracticeDocumento16 pagineIntroduction To Investigators Responsibilities With Good Clinical PracticeLuz Estefany Huaman MachacaNessuna valutazione finora
- A Comprehensive and Practical Guide to Clinical TrialsDa EverandA Comprehensive and Practical Guide to Clinical TrialsValutazione: 3 su 5 stelle3/5 (1)
- IRB Definitions (Is It Research? and Definitions of Exempt, Expedited and Full)Documento4 pagineIRB Definitions (Is It Research? and Definitions of Exempt, Expedited and Full)analyn123Nessuna valutazione finora
- Legal and Ethical Issues in ResearchDocumento5 pagineLegal and Ethical Issues in ResearchLiza3127Nessuna valutazione finora
- Chapter 14Documento15 pagineChapter 14Oneng IfayaniNessuna valutazione finora
- The Institutional Review Board: Monica B. Spaulding, M.D. Chair-Health Sciences IRB Professor of MedicineDocumento31 pagineThe Institutional Review Board: Monica B. Spaulding, M.D. Chair-Health Sciences IRB Professor of MedicineJennifer AlstonNessuna valutazione finora
- Research Ethics - Pub V2Documento32 pagineResearch Ethics - Pub V2nuhmanaljafriNessuna valutazione finora
- Ethical Issues PowerpointDocumento60 pagineEthical Issues PowerpointBilawal MughalNessuna valutazione finora
- 08 - Responsibilities of The Research Team WebinarDocumento43 pagine08 - Responsibilities of The Research Team WebinarVV28051986VNessuna valutazione finora
- Institutional Ethics Committee NotesDocumento38 pagineInstitutional Ethics Committee NotesAbhishek Nahar100% (1)
- HBM ExecDocumento11 pagineHBM ExecbleizherNessuna valutazione finora
- Curs GCP: What Is Good Clinical Practice?Documento6 pagineCurs GCP: What Is Good Clinical Practice?Delia NituNessuna valutazione finora
- Institutional Review Board/Independent Ethics Committee (Irb/Iec)Documento40 pagineInstitutional Review Board/Independent Ethics Committee (Irb/Iec)Swati chauhanNessuna valutazione finora
- Ethical and Legal Issues in Research Involving Human Subjects: Do You Want A Piece of Me?Documento5 pagineEthical and Legal Issues in Research Involving Human Subjects: Do You Want A Piece of Me?HugoMarcelo77Nessuna valutazione finora
- Clinical Research Definitions GuideDocumento56 pagineClinical Research Definitions GuideshyamkattiNessuna valutazione finora
- RESEARCH ETHICS & SCIENTIFIC MISCONDUCTDocumento30 pagineRESEARCH ETHICS & SCIENTIFIC MISCONDUCTPrabhu Prasad DevNessuna valutazione finora
- Good Epidemiological Practice (GEP) IEADocumento7 pagineGood Epidemiological Practice (GEP) IEAMarian MarchioriNessuna valutazione finora
- RHSC 500 Intro to Quantitative ResearchDocumento81 pagineRHSC 500 Intro to Quantitative ResearchOmeet HannahNessuna valutazione finora
- Part 1: Introduction Part 2: Responsibilities by Role Part 3: Summary of Key PointsDocumento13 paginePart 1: Introduction Part 2: Responsibilities by Role Part 3: Summary of Key PointsFaye BaliloNessuna valutazione finora
- Lectura 4Documento8 pagineLectura 4Marissella GutierrezNessuna valutazione finora
- What Special Protections Are Required To Enable Vulnerable Populations To Participate in Research?Documento63 pagineWhat Special Protections Are Required To Enable Vulnerable Populations To Participate in Research?Josue ColónNessuna valutazione finora
- IRB Review Protects Human SubjectsDocumento65 pagineIRB Review Protects Human SubjectsClaudia Quiñones RamosNessuna valutazione finora
- Role of The ErcDocumento97 pagineRole of The Ercaben101781Nessuna valutazione finora
- NCHRE - Aug 07Documento68 pagineNCHRE - Aug 07Igwe Emmanuel MichaelNessuna valutazione finora
- Introduction To EthicsDocumento11 pagineIntroduction To EthicsNurun NabiNessuna valutazione finora
- Dr. Josef Mengele's Inhumane Medical ExperimentsDocumento33 pagineDr. Josef Mengele's Inhumane Medical ExperimentsAngelaTrinidadNessuna valutazione finora
- ICMR Code: India's Guidelines for Ethical Biomedical ResearchDocumento4 pagineICMR Code: India's Guidelines for Ethical Biomedical ResearchSreeraj Guruvayoor SNessuna valutazione finora
- Kuliah KEPK 2015 Prof. FirmanDocumento203 pagineKuliah KEPK 2015 Prof. FirmanDian WijayantiNessuna valutazione finora
- Ethical Research Principles Protect SubjectsDocumento66 pagineEthical Research Principles Protect SubjectsTahany El BannaNessuna valutazione finora
- Clinical Trial ProcessDocumento16 pagineClinical Trial ProcessMohammed HammedNessuna valutazione finora
- University of Leicester Research Ethics Code of Practice: December 2006 IDocumento10 pagineUniversity of Leicester Research Ethics Code of Practice: December 2006 Istxavier5000Nessuna valutazione finora
- Research Ethics (Aanand) FinalDocumento11 pagineResearch Ethics (Aanand) FinalNaman DadhichNessuna valutazione finora
- Rev GCP Goals Principles Roles Resp Lepay Toth-Allen FDADocumento37 pagineRev GCP Goals Principles Roles Resp Lepay Toth-Allen FDASuhaimi JaaffarNessuna valutazione finora
- 2ahandout ENG 2020 ElearningDocumento5 pagine2ahandout ENG 2020 ElearningNupura AjeshNessuna valutazione finora
- (UWTSD) Research Ethics Integrity Code of Practice (2015-2018)Documento17 pagine(UWTSD) Research Ethics Integrity Code of Practice (2015-2018)Luis Rafael FajardoNessuna valutazione finora
- Ood Linical Ractice (GCP) in Clinical Research: Food and Drug AdministrationDocumento127 pagineOod Linical Ractice (GCP) in Clinical Research: Food and Drug AdministrationPap PipNessuna valutazione finora
- Increasing value and reducing waste in biomedical researchDocumento10 pagineIncreasing value and reducing waste in biomedical researchSamuel Andrés AriasNessuna valutazione finora
- Entrepreneurship and EconomicDocumento2 pagineEntrepreneurship and EconomicSukruti BajajNessuna valutazione finora
- Shri Siddheshwar Co-Operative BankDocumento11 pagineShri Siddheshwar Co-Operative BankPrabhu Mandewali50% (2)
- Structures Module 3 Notes FullDocumento273 pagineStructures Module 3 Notes Fulljohnmunjuga50Nessuna valutazione finora
- 10 Appendix RS Means Assemblies Cost EstimationDocumento12 pagine10 Appendix RS Means Assemblies Cost Estimationshahbazi.amir15Nessuna valutazione finora
- fr1177e-MOTOR CUMMINS 195HPDocumento2 paginefr1177e-MOTOR CUMMINS 195HPwilfredo rodriguezNessuna valutazione finora
- Reynold's Number Flow ExperimentDocumento8 pagineReynold's Number Flow ExperimentPatrick GatelaNessuna valutazione finora
- Mesa de Trabajo 1Documento1 paginaMesa de Trabajo 1iamtheonionboiNessuna valutazione finora
- ECED Lab ReportDocumento18 pagineECED Lab ReportAvni GuptaNessuna valutazione finora
- Sierra Wireless AirPrimeDocumento2 pagineSierra Wireless AirPrimeAminullah -Nessuna valutazione finora
- FZ16 9B 1KD2 (Patada) PDFDocumento62 pagineFZ16 9B 1KD2 (Patada) PDFPanthukalathil Ram100% (1)
- FC Bayern Munich Marketing PlanDocumento12 pagineFC Bayern Munich Marketing PlanMateo Herrera VanegasNessuna valutazione finora
- Harry Styles: The Rise of a Pop StarDocumento9 pagineHarry Styles: The Rise of a Pop StarBilqis LaudyaNessuna valutazione finora
- Pass Issuance Receipt: Now You Can Also Buy This Pass On Paytm AppDocumento1 paginaPass Issuance Receipt: Now You Can Also Buy This Pass On Paytm AppAnoop SharmaNessuna valutazione finora
- JRC Wind Energy Status Report 2016 EditionDocumento62 pagineJRC Wind Energy Status Report 2016 EditionByambaa BattulgaNessuna valutazione finora
- Anomaly Sell Out Remap December 2019 S SUMATRA & JAMBIDocumento143 pagineAnomaly Sell Out Remap December 2019 S SUMATRA & JAMBITeteh Nha' DwieNessuna valutazione finora
- Brightline Guiding PrinciplesDocumento16 pagineBrightline Guiding PrinciplesdjozinNessuna valutazione finora
- Assignment 2 - p1 p2 p3Documento16 pagineAssignment 2 - p1 p2 p3api-31192579150% (2)
- Telangana Budget 2014-2015 Full TextDocumento28 pagineTelangana Budget 2014-2015 Full TextRavi Krishna MettaNessuna valutazione finora
- (Jf613e) CVT Renault-Nissan PDFDocumento4 pagine(Jf613e) CVT Renault-Nissan PDFJhoanny RodríguezNessuna valutazione finora
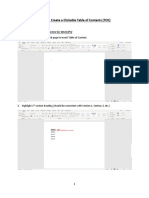
- How To: Create A Clickable Table of Contents (TOC)Documento10 pagineHow To: Create A Clickable Table of Contents (TOC)Xuan Mai Nguyen ThiNessuna valutazione finora
- 2023 Prospectus 2Documento69 pagine2023 Prospectus 2miclau1123Nessuna valutazione finora
- Nucleic Acid Isolation System: MolecularDocumento6 pagineNucleic Acid Isolation System: MolecularWarung Sehat Sukahati100% (1)
- Case Study Infrastructure ProjectsDocumento1 paginaCase Study Infrastructure ProjectsAnton_Young_1962Nessuna valutazione finora
- AssDocumento9 pagineAssJane SalvanNessuna valutazione finora
- Acknowledgment: George & Also To Our Group Guide Asst. Prof. Simy M Baby, For Their Valuable Guidance and HelpDocumento50 pagineAcknowledgment: George & Also To Our Group Guide Asst. Prof. Simy M Baby, For Their Valuable Guidance and HelpKhurram ShahzadNessuna valutazione finora