Documenti di Didattica
Documenti di Professioni
Documenti di Cultura
Chemioinformatica
Caricato da
Vicky TLCopyright
Formati disponibili
Condividi questo documento
Condividi o incorpora il documento
Hai trovato utile questo documento?
Questo contenuto è inappropriato?
Segnala questo documentoCopyright:
Formati disponibili
Chemioinformatica
Caricato da
Vicky TLCopyright:
Formati disponibili
Relazioni quantitative struttura-attività
Una relazione quantitativa struttura-attività (QSAR, Quantitative
Structure-Activity Relationship) mette in relazione una proprietà
delle struttura molecolare e la sua attività tramite un modello
matematico.
Viene utilizzato anche il termine "relazione quantitativa struttura-
proprietà" (QSPR) specialmente quando si prende in considerazione
una proprietà che non sia l'attività biologica.
Nelle progettazione di farmaci i metodi QSAR sono stati spesso
utilizzati per valutare le qualità dei farmaci al di là della loro
potenza in vitro.
Il più potente inibitore enzimatico (farmaco-dinamica) è di poca
utilità se incapace di raggiungere il propri bersagli (farmaco-
cinetica). L'attività in vivo di una molecola è composta spesso da
molti fattori. Uno studio struttura-attività può aiutare:
- a decidere quali sono le caratteristiche di una molecola che danno
origine alla sua attività
- a costruire composti modificati che abbiano proprietà amplificate.
- ad ottimizzare le risorse per fare i test riducendo al minimo i
tentativi (individuare le similitudini)
- a fare stime sulle proprietà di specie che non sono ancora state
testate
La relazione fra le proprietà numeriche derivate dalla struttura
(descrittori) e l'attività si può scrivere come una equazione dalla
forma generale:
attività=f(proprietà)
Definizione di attività biologica
La gran parte dei farmaci, per indurre una risposta deve interagire
fisicamente (cioè legarsi) con uno specifico bersaglio molecolare.
In un modello generale di azione di un farmaco sono necessarie 3
fasi per provocare una risposta biologica:
1) il farmaco (X) viene trasportato passivamente nelle vicinanze di
uno specifico recettore (R), cioè viene indirizzato verso qualche
regione esterna quindi, attraverso un "random walk", un numero
efficace di molecole riesce ad attraversare numerose membrane fino
ad arrivare nella zona bersaglio
R X
avvicinamento
2) si forma un complesso farmaco-recettore
Keq
R X
RX
3) il complesso formatosi può dar luogo a reazioni chimiche (come la
formazione di un legame covalente) o a cambiamenti conformazionali:
RX k
vel
RX *
Assimo Maris - Chemioinformatica 2011/12 1
Per ciascun caso particolare, non tutta questa sequenza è
necessariamente critica o limitante. Infatti, per la maggioranza
degli agenti farmaco-dinamici, il recettore non subisce alcuna
trasformazione chimica e la seconda fase determina la risposta
biologica.
D'altro canto, per molti agenti chemioterapici, la terza fase è
necessaria; per esempio la penicillina deve acilare il suo
bersaglio, l'enzima transpeptidasi dei batteri. Inoltre,
confrontando l'attività anti-batterica di una serie di derivati
penicillamici, sebbene la fase tre sia necessaria, potrebbe non
essere limitante poiché tutti i derivati potrebbero risultare
ugualmente efficaci nella reazione di acilazione del recettore, ma
differire nella capacità di attraversare la parete cellulare
batterica per raggiungere la zona bersaglio
La risposta biologica di una molecola può essere messa in relazione
con la probabilità “p” che una molecola del farmaco passi attraverso
tutte le tre fasi, ed è connessa alle probabilità p1, p2, p3 di
superare ogni singola fase dalla relazione:
p = p1 p2 p3
Se [X] è la concentrazione molecolare del farmaco si può dir che la
risposta è pari a:
risposta = costante [X] p
ovvero:
risposta = costante [X] p1 p2 p3
Nell'ambito ricerca farmacologica è naturale misurare, se possibile,
la concentrazione necessaria ad una risposta costante, per esempio:
- la EC50 (half maximal effective concentration) ovvero con la
concentrazione di un dato farmaco tale da produrre il 50%
dell'effetto massimale
- la IC50 (half maximal inhibitory concentration) ovvero la
concentrazione di un inibitore enzimatico necessaria per inibire il
50% dell'enzima.
- la LC50 (half maximal lethal concentration) ovvero la
concentrazione di una sostanza in grado di uccidere il 50% dei
soggetti
- la TC50 (half maximal toxic concentration) ovvero la concentrazione
di una sostanza che mostra un effetto tossico sul 50% dei soggetti
In ambito medico si parla in genere di dose (per es. LD50) piuttosto
che di concentrazione di farmaco. La relazione fra dose e
concentrazione è diretta se si assume che la concentrazione di
farmaco che raggiunge il recettore sia proporzionale alla dose
somministrata.
Considerando quindi la risposta come costante, la precedente
equazione può essere riscritta come:
1 costante
EC 50 p1 p 2 p 3
[ X ] risposta
Assimo Maris - Chemioinformatica 2011/12 2
L'uso dei logaritmi permette di linearizzare l'equazione che
diventa:
1 costante
log log log p1 log p 2 log p3
[X ] risposta
costante
Poiché il termine log è una costante si può scrivere:
risposta
1
attività log k log p1 log p2 log p3
[X ]
In questa formula l'attività del farmaco (il termine log 1/[X])
viene espressa come combinazione lineare di termini relativi al
trasporto del farmaco (log p1), all'attività intrinseca (log p2) e ad
eventuali trasformazioni del complesso farmaco-recettore (log p3).
Abbiamo visto in precedenza come gli effetti legati al trasporto
possano essere collegati al coefficiente di ripartizione
ottanolo/acqua.
Appare intuitivo collegare la probabilità che si formi un complesso
farmaco-recettore (p2) alla costante di equilibrio della reazione di
formazione e collegare la probabilità di trasformazione del
complesso alla costante di velocità di reazione corrispondente:
1
attività log k log p1 k2 log K eq k3 log k vel
[X ]
Se un farmaco si trova in prossimità del recettore, come nel caso di
un saggio in vitro, e se il recettore non subisce alcuna
trasformazione chimica, allora l'attività dipende solo dalla
reazione di formazione del complesso farmaco-recettore e l'equazione
si riduce a:
1
attività log k k2 log K eq
[X ]
Attività intrinseca
L’interazione farmaco-recettore può essere schematizzata nel modo
seguente:
k1
R X RX
k 1
Secondo questa ipotesi classica, il complesso farmaco-recettore RX è
l’unica entità in grado di iniziare la serie di eventi che porta
all’effetto finale, mentre R o X da soli sono inattivi.
L’interazione farmaco recettore è mediata da legami a bassa energia:
legami ionici, ponti idrogeno, attrazioni di van der Waals,
interazioni idrofobiche. Affinché il contatto tra il farmaco ed il
suo recettore persista per un tempo sufficiente a generare una
risposta biologica, occorre che il numero di legami a bassa energia
sia elevato, e che la superficie della molecola di farmaco e quella
di recettore siano chimicamente complementari l’una all’altra.
L’interazione farmaco-recettore segue la legge di azione di massa,
ovvero la velocità di reazione chimica è proporzionale al prodotto
delle concentrazioni dei reagenti.
Assimo Maris - Chemioinformatica 2011/12 3
Si definisce la costante di associazione o di affinità
k [ RX ]
Ka 1 che indica quanto la reazione è spostata verso la
k 1 [ R ] [ X ]
formazione del complesso ed è pertanto correlata alla forza del
legame chimico fra il farmaco ed il recettore.
Per tradizione però negli studi di binding si usa più frequentemente
la costante di equilibrio della reazione inversa, cioè di
dissociazione del complesso:
[ R] [ X ] 1
Kd
[ RX ] Ka
correlata inversamente all’affinità del ligando per il recettore.
La concentrazione totale dei recettori è data da:
[Rtot] = [R] + [RX]
se esprimiamo la concentrazione di recettori liberi in funzione di
Kd:
[ R] [ X ] K [ RX ]
Kd [ R] d
[ RX ] [X ]
e andiamo a sostituire si ottiene:
K d [ RX ] K
[ Rtot ] [ R] [ RX ] [ RX ] [ RX ] d 1
[X ] [X ]
Arrangiando questa ultima equazione, si ottiene l’equazione
fondamentale che correla la concentrazione di farmaco alla
concentrazione del complesso farmaco recettore:
[X ]
[ RX ] [ Rtot ]
Kd [X ]
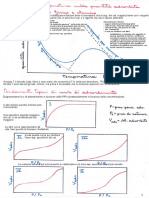
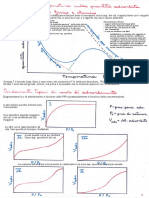
Graficamente questa equazione è rappresentata da un’iperbole rettangolare (in caso di scala
aritmetica), oppure da una curva sigmoide (in caso di scala semi-logaritmica).
[RX] [RX]
efficacia
[Rtot ] [Rtot ]
[Rtot ]/2 [Rtot ]/2
potenza (EC50)
[X] logEC50 log[X]
[X]=Kd
Scala lineare Scala semi-logaritmica
Sull’asse delle ascisse sono riportati i valori di concentrazione
del farmaco X, mentre sulle asse delle ordinate i valori di
concentrazione del complesso RX. Nel grafico, Rtot corrisponde
all’asintoto a cui la curva tende (plateau superiore alla curva),
Assimo Maris - Chemioinformatica 2011/12 4
mentre [X]=Kd corrisponde alla concentrazione del farmaco X
necessaria per saturare il 50% di tutti i siti presenti.
[X ] X ] K d Kd [R ]
[ RX ] [ Rtot ] [ [ RX ] [ Rtot ] tot
Kd [X ] Kd Kd 2
Possiamo quindi identificare la Kd con la EC50
Poiché la costante di dissociazione Kd è determinata dalla differenza
di energia libera fra lo stato legato e quello non legato possiamo
scrivere:
1
G d0 RT ln K d 2.3RT log K d 2.3RT log 2.3RT log K a 2.3RT pK d
Kd
infine, sostituendo Kd con EC50:
Gd0 2.3RT log EC 50 2.3RT pEC50
Equazione di Hammett
Hammett (1930) ha dato un contributo decisivo alla correlazione fra
struttura e proprietà, trovando un modello con il quale è possibile
prevedere alcune costanti chimico-fisiche e il comportamento chimico
alla variazione di energia libera indotta da determinati sostituenti
nella struttura di riferimento.
Era già noto che la forza acida, misurata dalla costante di
dissociazione Kd, dell’acido benzoico può essere più o meno
sensibilmente influenzata da sostituenti: sostituenti che attirano
elettroni stabilizzano l’anione carbossilato spostando l’equilibrio
verso destra e quindi aumentando la forza acida, viceversa i
sostituenti elettron-repulsori.
- +
COOH COO + H
X X
Hammett ha rappresentato la forza acida dell’acido benzoico e la
forza acida di un acido benzoico para- (o meta-) sostituito con
l'equazione di una retta:
log K H H o anche log kH=H
log K X X o anche log kX=X
dove:
- K è la costante di equilibrio della reazione
- k è la costante di velocità della reazione
- è una costante caratteristica della reazione o equilibrio in
oggetto e che dipende dalle condizioni sperimentali (temperatura,
solvente, ...)
- è il cosiddetto parametro di sostituzione di Hammet che misura
l’effetto elettronico del sostituente e deve dipendere solamente dal
tipo di sostituente e dalla sua posizione (meta- o para-).
Sottraendo dalla seconda la prima equazione si ha:
Assimo Maris - Chemioinformatica 2011/12 5
log K X log K H X H
L'equazione si può rielaborare come:
KX
log X H
KH
o anche:
log K X X H log K H
Ponendo uguale a zero l’effetto elettromerico dell’idrogeno, cioè
del termine di riferimento, il termine -H si annulla, mentre il
termine log KH, che è costante all’interno di tutta la serie di acidi
benzoici sostituiti, può essere sostituito dalla costante “c”,
quindi allo stesso modo di dipende dal tipo di acido di partenza e
dalle condizioni sperimentali:
log K X X c
La cosa interessante è che con lo stesso parametro elettronico X,
una volta determinata sperimentalmente la costante di reazione per
una nuova serie, si può prevedere con discreta approssimazione la
forza acida tutti gli acidi aromatici.
Diagrammando il log KX in funzione di X si ottiene una retta dove
rappresenta la pendenza e c l’intercetta. Quanto maggiore è la
pendenza, tanto maggiore è la sensibilità della reazione agli
effetti elettronici.
I parametri si trovano i tabelle di dati chimico-fisici.
L’equazione log K X X c è detta anche di tipo LFER (Linear Free
Energy Relation) in quanto il logaritmo della costante di equilibrio
Assimo Maris - Chemioinformatica 2011/12 6
di una reazione è proporzionale all’energia libera della reazione
stessa:
GX = -RTln KX
ed è quindi possibile esprimere l’energia libera stessa tramite una
relazione lineare:
GX = -2.3RTlog KX
GX = -2.3RT(+c)
GX = (-2.3RT)X – (2.3RTc)
GX = ’X – c’
Dove ’ e c’ sono delle costanti e il parametro sX misura la
variazione di energia libera causata dalla presenza del sostituente.
Si può anche prevedere la velocità di reazioni nelle quali il
fattore elettronico gioca un ruolo determinante.
Ad esempio alla velocità di idrolisi degli esteri degli acidi
carbossilici è applicabile con buona approssimazione l’equazione di
Hammett, logicamente dopo aver trovato sperimentalmente su alcuni
termini il valore ottimale da assegnare a ; mentre al posto di c si
pone il logaritmo della velocità d’idrolisi dell’estere non
sostituito.
Tuttavia nei sistemi aromatici gli effetti elettronici, induttivi e
mesomerici, dipendono dalla posizione del sostituente; cosicché
bisogna adoperare valori diversi di per le sostituzioni in para e
per quelle in meta.
Con la serie orto-sostituita non è più possibile ottenere una retta
con gli stessi valori di X e con gli opportuni valori di , perché
in questo caso assumono importanza non trascurabile effetti diversi
da quello elettronico, come compressione sterica, chelazione, ecc.
Inoltre i valori di X calcolati per la serie aromatica non sono
validi per la serie alifatica, dove gli effetti mesomerici non sono
più presenti; e all’interno della serie alifatica stessa cambiano
con la distanza del sostituente dal gruppo funzionale di
riferimento.
Possiamo utilizzare la relazione che abbiamo determinato per la
costante di equilibrio ( log K X X c ) per sostituire il termine
log K eq nell'equazione della attività intrinseca:
1
attività log k k 2 log K eq
[X ]
1
attività log k k2 X c
[X ]
o più in generale:
1
attività log k ' k 2 ' X
[X ]
Assimo Maris - Chemioinformatica 2011/12 7
Dove k’ rappresenta l’attività biologica del composto di riferimento
o l’ attività media dei composti della serie.
Questa equazione è predittiva perché dalla conoscenza dei parametri
X si può valutare l’attività, ma anche diagnostica perché ogni
composto che obbedisce alla suddetta equazione agisce probabilmente
secondo lo stesso meccanismo.
Esempio: effetto antibatterico dei sulfamidici
p-NH2-C6H4-SO2-NH-C6H4-R
Utilizzando un insieme di 17 molecole è stato parametrizzato con X
l’effetto del sostituente R sul gruppo solfonammidico ottenendo la
relazione:
attività = 1.05 R - 1.28 r=0.97
Questa relazione indica che il sostituente R non si lega al
recettore, ma influenza l’attività dolo in quanto influisce sulla
ionizzazione del composto. Questo risultato è in accordo con l’ormai
accettato meccanismo di azione dei sulfamidici, cioè come
antagonisti dell’anione dell’acido para-amminobenzoico (p-NH2-C6H4-
COO-). La reazione critica è la ionizzazione nel mezzo biologico.
Vi sono pochi esempi in cui un solo parametro, anche se di
fondamentale importanza nell’interazione farmaco-organismo, rende
conto di tutta la variazione dei dati di attività e, in generale è
necessario tenere conto anche di altri parametri.
Hansch (1964) per primo ha introdotto un modello QSAR dove
l’attività è data dalla sommatoria del contributo di diversi fattori
chimico-fisici.
Per una serie di composti analoghi ha proposto la seguente
relazione:
1
log c i ki Fi
[X ]
dove i termini Fi sono le proprietà considerate e i termini ki sono i
relativi coefficienti.
In un tipico esempio concreto l’equazione può assumere la seguente
forma:
1
log k1 k 2 2 k3 k 4 ES k5 MR ... c
[X ]
dove:
ki i termini ki sono valori costanti che dipendono dal tipo di
attività e dalla serie di composti considerata.
c c è l'attività del composto non sostituito o di
riferimento, infatti quando tutti i ki sono nulli, allora
1
log c
[X ]
PX è la costante idrofobica del sostituente e rappresenta il
log
PH contributo di un dato gruppo (X) al carattere idrofobico,
Assimo Maris - Chemioinformatica 2011/12 8
ed è responsabile dell’assorbimento e della distribuzione.
PX e PH sono i coefficienti di ripartizione fra ottanolo e
acqua del composto sostituito e del composto di riferimento
rispettivamente.
è il fattore di Hammett e rappresenta l'effetto
elettronico del sostituente; è responsabile soprattutto del
legame con il recettore
ES ES è il fattore sterico
Il fattore sterico è stato valutato per ciascun gruppo
sostituente ‘X’ misurando la velocità relativa di idrolisi
acida di metilesteri di acidi acetici -sostituiti
(correggendo eventuali effetti induttivi o iper-coniugativi
del sostituente stesso):
Il fattore sterico si ricava dal rapporto della velocità di
kX
idrolisi delle due reazioni: E s log
kH
Il fattore sterico è stato calcolato da Verloop anche dai
raggi di Van der Waals opportunamente valutati:
B4
B3
L
B1 B2
L Lunghezza del gruppo lungo l’asse del legame
B1 Dimensione minima
B4 Dimensione massima
B2 , B3 dimensioni intermedie ortogonali ad L e fra di loro.
MR MR è la rifrazione molare.
La rifrazione molare ha le dimensioni di un volume e si
presta bene a correggere e convalidare Es e, talvolta,
anche il parametro idrofobico .
Da allora un enorme numero di equazioni QSAR è stato riportato in
letteratura e molti diversi parametri sono stati sviluppati per
rappresentare caratteristiche idrofobiche, elettroniche o steriche.
Le proprietà che si includono nelle equazioni QSAR devono essere fra
loro il meno correlate possibile.
In principio le QSAR furono derivate per insiemi di serie di
composti che differiscono fra loro solo per una parte della
molecola.
Assimo Maris - Chemioinformatica 2011/12 9
Tali differenze possono spesso essere caratterizzate usando
appropriate costanti per ogni sostituente, che sono pubblicate in
tabelle.
Un altro tipo di "parametro" che appare spesso nelle equazioni QSAR
è la variabile "indicator". Tali variabili servono ad estendere una
equazione QSAR a diversi tipi di serie di molecole, rendendole di
maggior applicabilità.
Per esempio Hansch e i suoi colleghi derivarono la seguente
equazione per le costanti di legame delle solfonammidi (X-C6H4-SO2-
NH2) alla anidrasi carbonica umana:
log K 0.64 log P 1.55 2.07 I1 3.28 I 2 6.94
I1 assume valore 1 per meta-sostituenti (0 per gli altri) e I2 è 1
per gli orto sostituenti (0 per gli altri).
Sono stati sviluppati anche dei modelli che invece di basarsi su
tali proprietà, si basano sulla struttura o caratteristiche
strutturali. Fondamentalmente la struttura è la somma di alcuni
componenti elementari (atomi, tipi di atomi) o caratteristiche
(connessioni di atomi) oppure è la definizione della probabilità di
una loro localizzazione.
I più semplici di tali modelli (Free-Wilson, 1964) consistono nel
confrontare variazioni di attività biologica all’interno di una
serie di molecole con scheletro molecolare in comune, che
differiscano per la presenza/assenza di particolari sostituenti o
frammenti. Si tratta poi di risolvere un sistema di equazioni del
tipo:
Attività i a j kij a j
dove a è l’attività del composto di riferimento o l’attività media
della serie e di kij è una “variabile indicatrice” che è pari a zero
se il frammento j-esimo è assente nella molecola i-esima o è pari a
uno se il frammento è presente, per trovare i migliori valori di aj
che è il contributo all’attività del frammento j-esimo.
Il vantaggio dell'approccio Free-Wilson è che la QSAR si determina
con un numero di dati minore, ma i valori che si ottengono in
diverse condizioni o con diversi recettori non sono in genere
confrontabili. Si noti inoltre che questo approccio si basa
sull’assunzione che il contributo di ogni sostituente sia costante e
additivo, indipendentemente dal suo intorno intra-molecolare.
Un metodo più rigoroso è quello della connettività molecolare,
sviluppato da Kier a Hall, che si fonda sulla topologia. Questo
metodo produce una serie di indici numerici che riflettono la
presenza nella molecola di caratteristiche strutturali come
ramificazioni, insaturazioni, cicli e posizioni di eteroatomi.
Questi indici sono unici, dipendono dall’intorno molecolare e
possono essere correlati a proprietà chimico-fisiche o biologiche-
Per un approccio ancora più rigoroso si ricorre al calcolo
quantomeccanica che permette di determinare la struttura molecolare
Assimo Maris - Chemioinformatica 2011/12 10
e l’energia relativa di diversi conformeri e che rappresenta gli
aspetti elettronici in termini di localizzazione degli elettroni.
La possibilità di poter descrivere le molecole in maniera così
accurata permette di derivare delle QSAR non solo per insiemi di
serie di composti che differiscono fra loro solo per una parte della
molecola.
Più di recente infatti l'analisi si è rivolta a serie non con-
generiche di composti, calcolando direttamente dei descrittori per
l'intera molecola.
Per esempio la cosiddetta "analisi della forma molecolare" include
descrittori che misurano la forma relativa dei composti:
1) si effettua una analisi conformazionale dei composti e si
individuano le strutture con minima energia
2) si sovrappongono tali conformazioni a una struttura di
riferimento (tipicamente uno dei composti più attivi della serie)
3) si calcolano il volume di sovrapposizione e di non
sovrapposizione che saranno i parametri da includere nell'equazione
QSAR.
Selezione di composti per una analisi QSAR
La derivazione di una equazione QSAR è costituita da un certo numero
di passi distinti.
Innanzitutto è necessario sintetizzare i composti e determinare la
loro attività biologica. Quando si decide quali composti
sintetizzare è importante coprire l'intervallo di proprietà che
possono avere influenza sull'attività. Questo significa applicare le
procedure di verifica e di manipolazione dei dati viste in
precedenza. Per esempio non ha senso considerare una serie di
composti che hanno coefficienti di ripartizione simili se si crede
che questa proprietà sia importante.
Per decidere quali composti sintetizzare per poter estrarre la
massima informazione possibile dal minor numero di molecole si può
ricorrere alle cosiddette "tecniche di progettazione
dell'esperimento".
Fra i vari metodi quello che si può capire in modo più diretto è il
"full factorial design".
Full factorial design
Supponiamo di avere 2 variabili (dette "fattori") che possono
influenzare il risultato (detto "risposta") di un esperimento.
In un esperimento chimico:
- i fattori potrebbero essere la temperatura e il pH
- la risposta potrebbe essere la resa del prodotto.
In un esperimento per lo studio dell'inibizione di un enzima:
- i fattori potrebbero essere il coefficiente di ripartizione
ottanolo/acqua della molecola e il parametro di Hammett per i
sostituenti
Assimo Maris - Chemioinformatica 2011/12 11
- la risposta potrebbe essere il grado di inibizione misurato come
IC50
Supponiamo inoltre di essere interessati a 2 soli valori per ogni
fattore (T1, T2, pH1, pH2), allora sono possibili 4 diversi
esperimenti:
(T1, pH1) (T1, pH2)
(T2, pH1) (T2, pH2)
I primi 3 esperimenti servono a misurare l'effetto del cambiamento
di una variabile alla volta, mentre il quarto esperimento misura
l'effetto del cambiamento di entrambe le variabili e permette di
identificare le possibili interazioni fra i fattori.
Se ci fossero 3 fattori, con 2 valori ognuno allora il "full
factorial design" consisterebbe in 2³=8 esperimenti e la possibilità
di esplorare le possibili interazioni fra 2 o 3 fattori
In genere i fattori singoli sono più importanti delle interazioni di
coppia, che loro volta sono più importanti delle interazioni triple
e così via.
Si può quindi decidere di ridurre il numero di esperimenti
utilizzando il cosiddetto "fractional factorial design".
Per esempio in un "half factorial design" il numero di esperimenti è
la metà rispetto al "full factorial design".
Ovviamente con un minore numero di esperimenti potrebbe risultare
meno immediato determinare quali sono i fattori, o le combinazioni
di fattori più importanti.
I metodi di progettazione fattoriale a volte non sono applicabili a
studi di tipo QSAR. Per esempio potrebbe non essere possibile
produrre tutti i composti che coprano tutte le combinazioni di
fattori previste.
In tal caso bisogna scegliere un sotto-insieme di molecole, fra
quelle disponibili, che ricopra il più ampio spettro possibile di
valori nello spazio delle variabili.
Un tecnica per effettuare tale selezione è il "D-optimal design":
- per ogni sottoinsieme di molecole possibile si costruisce una
matrice Dnp in cui ognuna delle n righe corrisponde a una molecola e
ognuna delle p colonne corrisponde a un descrittore.
descritt.1 ... descritt.p
n p n p
molecola1 x11 x1 ... x1 p x p d 11 ... d1 p
D n p ... ... x ji xi ... ... d ji ...
molecola n x n1 x1 ... x np x p d n1
... d np
dove x ji è il valore del descrittore i-esimo per la molecola j-esima,
n
xi x ji è il valore medio del descrittore i-esimo calcolato su tutte
j 1
le molecole e volendo utilizzare una notazione compatta, si
definisce d ji x ji xi .
Assimo Maris - Chemioinformatica 2011/12 12
- si costruisce la matrice di varianza-covarianza
Z p p
D T p n
D
n p
n
i cui elementi fuori diagonale rappresentano la covarianza fra due
variabili (piccola se i valori dei descrittori delle n molecole
considerate sono poco correlati):
d1 j
1 1 1 n 1 n
z ij d 1i ... d ni ... ( d1i d 1 j ... d ni d nj ) d ki d kj ( x ki xi ) ( x kj x j )
n n n k 1 n k 1
d nj
e i cui elementi sulla diagonale sono la varianza per ogni variabile
(grande se la dispersione dei valori dei descrittori delle n
molecole considerate è grande):
1 n 1 n
z jj d kj d kj ( xkj xk ) 2
n k 1 n k 1
(N.B. La matrice di "varianza-covarianza" Z è una matrice quadrata
simmetrica, per cui è diagonalizzabile.)
- si calcola il determinante della matrice di varianza-covarianza
che è tanto più grande quanto maggiore è la varianza e minore la
covarianza.
- si seleziona il sotto-insieme di molecole che massimizza tale
determinante.
Esempio con 3 molecole e 2 descrittori:
x11 x1 x12 x 2 d11 d12
D 3 2
x 21 x1 x 22 x 2 d 21 d 22
x31 x1 x32 x 2 d 31 d 32
d11 d 21 d 31
DT
d 12 d 22 d 32
d11 d12
d d 21 d 31
D D 11 d 21 d 22
d 32
T
d12 d 22
d 31 d 32
d d d 21 d 21 d 31 d 31 d11 d12 d 21 d 22 d 31 d 32
D T D 11 11
d12 d11 d 22 d 21 d 32 d 31 d12 d12 d 22 d 22 d 32 d 32
3 3
d k1 d
2
k1 dk 2
D T D 3 k 1 k 1
3
d d dk 2
2
k 1
k1 k2
k 1
Assimo Maris - Chemioinformatica 2011/12 13
3 2 3
d k1 d k1 dk 2
k 1 k 1
DT D
Z 3 n 3
n
n
d k1 d k 2 2
dk 2
k 1 k 1
n n
3 3 3 3
d k1 dk2 d k1 d k 2 d
2 2
k1 dk2
Z k 1
k 1
k 1
k 1
n n n n
2
3 3
3
d k1
2
dk2
2
d k1 d k 2
Z k 1
2
k 1
k 1 2
n n
Esempio numerico con 5 molecole e 3 descrittori:
1) matrice iniziale
p1 p2 p3
m1 3 2 3
m2 3 3 1
m3 2 2 2
m4 2 2 3
m5 1 1 1
2) calcolo il valor medio per ogni variabile
p1 p2 p3
2.2 2 2
3) sottraggo il valor medio a ogni descrittore
p1 p2 p3
m1 0.8 0 1
m2 0.8 1 -1
m3 -0.2 0 0
m4 -0.2 0 1
m5 -1.2 -1 -1
4) calcolo la matrice trasposta
m1 m2 m3 m4 m5
p1 0.8 0.8 -0.2 -0.2 -1.2
p2 0 1 0 0 -1
p3 1 -1 0 1 -1
5) moltiplico la matrice trasposta per la matrice stessa
p1 p2 p3
p1 2.8 2 1
p2 2 2 0
p3 1 0 4
6) divido per il numero di molecole, ovvero per 5
p1 p2 p3
p1 0.56 0.4 0.2
Assimo Maris - Chemioinformatica 2011/12 14
p2 0.4 0.4 0
p3 0.2 0 0.8
7) interpreto i dati
- lungo la diagonale si trovano i valori della varianza per ogni
descrittore. Il descrittore p3 ha la varianza più grande (0.8), e il
descrittore p2, la più piccola (0.4). Quindi possiamo dire che i
valori di p3 variano più dei valori di p2.
- la covarianza viene visualizzata negli elementi fuori dalla
diagonale della matrice. Sia la covarianza tra p1 e p2 (0.4) che la
covarianza tra la p1 e p3 sono positive (0.2), questo significa che
i valori tendono a covariare in modo positivo: quando p1 cresce
anche i valori di p2 e p3 tendono a salire, e viceversa. La
covarianza tra p2 e p3, invece, è pari a zero, ciò significa che non
c'è relazione tra le variazioni dei valori di p2 e di p3
- valori negativi di covarianza indicano che i valori tendono a
muoversi in direzioni opposte
Interpretazione di una equazione QSAR
Cosa si fa con una equazione QSAR una volta che è stata derivata?
Un uso ovvio è quello di prevedere le attività di molecole non
ancora determinate sperimentalmente, e possibilmente non ancora di
sintetizzate.
La capacità predittiva di una QSAR è generalmente più accurata per
previsioni di tipo interpolativo (vale a dire per i composti i cui
valori dei parametri ricadono all'interno dell'intervallo di quelle
considerate nell’insieme di dati) che per le previsioni
estrapolativo (composti che sono al di fuori dell'intervallo
considerato).
Una equazione QSAR può fornire spunti sul meccanismo del processo
che si sta studiando. Abbiamo già visto, per esempio come la
presenza di una relazione parabolica tra l'attività e il logaritmo
del coefficiente di ripartizione venga interpretata in termini di
trasporto di un composto fino al recettore.
Le QSAR sono spesso interpretate in termini di interazioni
specifiche con il bersaglio macromolecolare.
Nei casi in cui la struttura cristallina del complesso ligando-
recettore sia stata successivamente determinata è stato possibile
utilizzare la computer grafica molecolare per verificare se i
parametri dell'equazione QSAR avessero una qualche corrispondenza
"fisica".
Anche l'assenza di una correlazione può fornire indicazioni utili.
Per esempio, se una serie di parametri dà una correlazione migliore
di un'altra, allora questo può indicare che è in funzione un
meccanismo particolare.
Se per una serie di composti non vi è alcuna correlazione con un
parametro (ad esempio, una misura sterica) allora questo potrebbe
Assimo Maris - Chemioinformatica 2011/12 15
indicare che la proprietà associata (cioè il volume sterico) è di
scarsa importanza.
Assimo Maris - Chemioinformatica 2011/12 16
Potrebbero piacerti anche
- 2 2 Calcolo Aste ComposteDocumento7 pagine2 2 Calcolo Aste ComposteFrank LottNessuna valutazione finora
- Il Ponte Di WheatstoneDocumento11 pagineIl Ponte Di WheatstoneProject Symphony Collection100% (3)
- Push OverDocumento12 paginePush Overandrea_l1349Nessuna valutazione finora
- EnzimiDocumento13 pagineEnzimiAndrea_Gerosa_3807Nessuna valutazione finora
- Farmacologia GeneraleDocumento57 pagineFarmacologia Generalepattada3333100% (1)
- RecettoriDocumento36 pagineRecettoriAlessandro IsirdiNessuna valutazione finora
- Lezione 10 30-10 PT 1Documento6 pagineLezione 10 30-10 PT 1RivettiLucaNessuna valutazione finora
- MetodiDocumento49 pagineMetodiMarioNessuna valutazione finora
- Interazione Tra Farmaco e RecettoreDocumento7 pagineInterazione Tra Farmaco e RecettoreElena BeniNessuna valutazione finora
- Studio ChimicaDocumento3 pagineStudio ChimicaFrancesco CartaNessuna valutazione finora
- 2 - Teorie RecettorialiDocumento34 pagine2 - Teorie RecettorialiακηςNessuna valutazione finora
- Lezione Reattori ChimiciDocumento4 pagineLezione Reattori ChimiciAngelo Ngiò Francesco IovinoNessuna valutazione finora
- Capitolo 18 ChimicaDocumento3 pagineCapitolo 18 ChimicaSteven ZeneliNessuna valutazione finora
- EnzimiDocumento17 pagineEnzimibiagio castronovoNessuna valutazione finora
- Velocità Di ReazioneDocumento16 pagineVelocità Di ReazioneFrancesco TorrettaroNessuna valutazione finora
- Cinetica Chimica in Presenza Di Reazioni Multiple: Capitolo 6Documento26 pagineCinetica Chimica in Presenza Di Reazioni Multiple: Capitolo 6polaise_rockNessuna valutazione finora
- Chi Go 160 Cine Tica 1112Documento12 pagineChi Go 160 Cine Tica 1112Ivan Alberto NinaNessuna valutazione finora
- Bubu Settere23Documento7 pagineBubu Settere23Emanuele SantonastasoNessuna valutazione finora
- RisonanzaDocumento15 pagineRisonanzaEmanuele FormicaNessuna valutazione finora
- Come Agiscono I Farmaci Caratteristiche Del Legame Farmaco RecettoreDocumento83 pagineCome Agiscono I Farmaci Caratteristiche Del Legame Farmaco Recettoreapi-26155364Nessuna valutazione finora
- Catalisi OmogeneaDocumento20 pagineCatalisi OmogeneaLuca MarlettaNessuna valutazione finora
- Cinetica PDFDocumento10 pagineCinetica PDFvirgingitorige55Nessuna valutazione finora
- Lucidi 3°blocco (25 Nov)Documento49 pagineLucidi 3°blocco (25 Nov)progettoMTBENessuna valutazione finora
- Lucidi 3°bloccoDocumento49 pagineLucidi 3°bloccoprogettoMTBENessuna valutazione finora
- Farmacologia Pt. 2Documento29 pagineFarmacologia Pt. 2MADDALENA BARALDINessuna valutazione finora
- La Cinetica Chimica 2Documento49 pagineLa Cinetica Chimica 2Stefano MarroneNessuna valutazione finora
- NMR PDFDocumento41 pagineNMR PDFgiulia pacchiarotti100% (1)
- ImpedenziometriaDocumento5 pagineImpedenziometriaadrianaNessuna valutazione finora
- La Velocità Delle Reazioni ChimicheDocumento3 pagineLa Velocità Delle Reazioni Chimicheclasse 1 ANessuna valutazione finora
- Fisica Della MateriaDocumento36 pagineFisica Della MateriaRiccardo Della MonicaNessuna valutazione finora
- Lezione 10Documento10 pagineLezione 10Basette BasetteNessuna valutazione finora
- Organica Superiore (B J Kakos)Documento96 pagineOrganica Superiore (B J Kakos)Vassily KakosNessuna valutazione finora
- Farmacodinamica SettembreDocumento30 pagineFarmacodinamica SettembreChiaraPetraroliNessuna valutazione finora
- FARMACODINAMICADocumento3 pagineFARMACODINAMICAMarta DonghiNessuna valutazione finora
- Circuiti RLC in RisonanzaDocumento4 pagineCircuiti RLC in RisonanzaEttore D'AgostinoNessuna valutazione finora
- Cinetica QuimicaDocumento15 pagineCinetica Quimicajivifer14Nessuna valutazione finora
- Int Non Covalenti PDFDocumento19 pagineInt Non Covalenti PDFEsterino VaccaNessuna valutazione finora
- Dosaggio EnzimaticoDocumento6 pagineDosaggio EnzimaticoBabumba92Nessuna valutazione finora
- Fisiopatologia Lezione 3Documento4 pagineFisiopatologia Lezione 3Mariagiovanna MariagiovannaNessuna valutazione finora
- Lezione1 Ott02Documento20 pagineLezione1 Ott02api-26155364Nessuna valutazione finora
- 13 MonteCarloDocumento8 pagine13 MonteCarlodon-donnolaNessuna valutazione finora
- Farmacologia Lez2Documento7 pagineFarmacologia Lez2Mariarita Di BenedettoNessuna valutazione finora
- Elettroanalitica 11 - Voltammetria Parte 5a - Volt CiclicaDocumento28 pagineElettroanalitica 11 - Voltammetria Parte 5a - Volt CiclicaJoey ShortinoNessuna valutazione finora
- Parametri Modali n1 PDFDocumento20 pagineParametri Modali n1 PDFAlessio ScarabelliNessuna valutazione finora
- Farma 3Documento4 pagineFarma 3Mariarita Di BenedettoNessuna valutazione finora
- Dynamic Light ScatteringDocumento6 pagineDynamic Light ScatteringFederico AmatoNessuna valutazione finora
- Cinetica ChimicaDocumento3 pagineCinetica ChimicaracsanvalentinaNessuna valutazione finora
- Interpretasi DataDocumento3 pagineInterpretasi DataARESMA SUSILAWATINessuna valutazione finora
- Reattore ContinuoDocumento6 pagineReattore Continuodavidedamiani5Nessuna valutazione finora
- Ilovepdf Merged (3Documento32 pagineIlovepdf Merged (3Pierluigi LibertiNessuna valutazione finora
- Relazione Tra Campo Elettrostatico e PotenzialeDocumento2 pagineRelazione Tra Campo Elettrostatico e Potenzialealessandro temelliniNessuna valutazione finora
- Parte 2 Macromolecolare2Documento34 pagineParte 2 Macromolecolare2Iolanda PucaNessuna valutazione finora
- Quaderno - Elettronica Nicola Auricchio PDFDocumento292 pagineQuaderno - Elettronica Nicola Auricchio PDFYuriAmbrosio100% (1)
- Trasferimento Di MateriaDocumento56 pagineTrasferimento Di Materianp484Nessuna valutazione finora
- 5elettronica gvp2Documento41 pagine5elettronica gvp2Artem KubantsevNessuna valutazione finora
- 7 ReattoriDocumento25 pagine7 ReattoriFrancesco FuscoNessuna valutazione finora
- HartreeDocumento12 pagineHartreeioenopioNessuna valutazione finora
- 2 CPB Odontoiatria 2019Documento48 pagine2 CPB Odontoiatria 2019Maria MigliettaNessuna valutazione finora
- Esercizi di matematica: equazioni integrali e integro-differenzialiDa EverandEsercizi di matematica: equazioni integrali e integro-differenzialiNessuna valutazione finora
- Esercizi di matematica: calcolo tensorialeDa EverandEsercizi di matematica: calcolo tensorialeValutazione: 5 su 5 stelle5/5 (1)
- Analisi Sperimentale Di Collegamenti Trave-Colonna Equipaggiati Con Dissipatori Ad Attrito e Progettazione Di Edificio P 1 PDFDocumento314 pagineAnalisi Sperimentale Di Collegamenti Trave-Colonna Equipaggiati Con Dissipatori Ad Attrito e Progettazione Di Edificio P 1 PDFVincenzo PilusoNessuna valutazione finora
- DYWIDAG - Ancoraggi A BarreDocumento8 pagineDYWIDAG - Ancoraggi A BarreClaudioNessuna valutazione finora
- Prove Pressiometriche Menard PDFDocumento43 pagineProve Pressiometriche Menard PDFccostrutNessuna valutazione finora