Potrebbero piacerti anche
- Chimica Organica AppuntiDocumento32 pagineChimica Organica Appuntity rellandoNessuna valutazione finora
- Derivati Degli Acidi Carbossilici (Schemi e Meccanismi) - Chimica Organica.Documento21 pagineDerivati Degli Acidi Carbossilici (Schemi e Meccanismi) - Chimica Organica.Giorgio MattaNessuna valutazione finora
- Appunti Di Chimica 2Documento6 pagineAppunti Di Chimica 2valerio.vollaroNessuna valutazione finora
- Dispensa Acidi CarbossiliciDocumento20 pagineDispensa Acidi CarbossiliciFrancesco BerniniNessuna valutazione finora
- Esercizi Chimica OrganicaDocumento108 pagineEsercizi Chimica OrganicaminchiazzurraNessuna valutazione finora
- Aldeidi e Chetoni (10 e 11 Maggio)Documento8 pagineAldeidi e Chetoni (10 e 11 Maggio)Hi HiNessuna valutazione finora
- Gruppi Funzionali, Polimeri e CarboidratiDocumento10 pagineGruppi Funzionali, Polimeri e CarboidratiLuca CanegratiNessuna valutazione finora
- Condensazione AldolicaDocumento23 pagineCondensazione AldolicaAnonymous Dngi7UNessuna valutazione finora
- Mat Did 334176Documento45 pagineMat Did 334176Miguel RamírezNessuna valutazione finora
- C. Organica COMPLETODocumento18 pagineC. Organica COMPLETOAndra IoanaNessuna valutazione finora
- Reazioni AldeidiDocumento20 pagineReazioni AldeidistvlnpllNessuna valutazione finora
- Alcani, Alcheni, AlchiniDocumento19 pagineAlcani, Alcheni, AlchiniMassimiliano CameroniNessuna valutazione finora
- Breve Descrizione Reazioni Chimica OrganicaDocumento16 pagineBreve Descrizione Reazioni Chimica OrganicaAlessio MenegonNessuna valutazione finora
- Chimica Organica EsamiDocumento18 pagineChimica Organica Esamity rellandoNessuna valutazione finora
- 20 - Reattività Del Carbonio in Alfa Al CarbonileDocumento18 pagine20 - Reattività Del Carbonio in Alfa Al CarbonileAldo BlesiNessuna valutazione finora
- Aldeidi e ChetoniDocumento21 pagineAldeidi e ChetonimikadoboomNessuna valutazione finora
- Alogeno DerivatiDocumento7 pagineAlogeno Derivatigra_zianoNessuna valutazione finora
- UntitledDocumento21 pagineUntitledapi-253266324Nessuna valutazione finora
- Lezione 33 Chimica - YMP1 - Classe 1 - 1 - Allegato - 1Documento42 pagineLezione 33 Chimica - YMP1 - Classe 1 - 1 - Allegato - 1Francesco CarrozzoNessuna valutazione finora
- 2.1 Dai Gruppi Funzionali Ai PolimeriDocumento2 pagine2.1 Dai Gruppi Funzionali Ai PolimeriMarino MariniNessuna valutazione finora
- Chimica - Reazioni Di Organica by SilvioDocumento12 pagineChimica - Reazioni Di Organica by SilvioIvanoBarnabaNessuna valutazione finora
- Programma Chimica OrganicaDocumento2 pagineProgramma Chimica OrganicaSimon CarrinoNessuna valutazione finora
- Nannai - Reazioni Degli AlcaniDocumento13 pagineNannai - Reazioni Degli AlcaniNannai02Nessuna valutazione finora
- ALCHINIDocumento17 pagineALCHINIGiuseppe ReinaNessuna valutazione finora
- Acidi CarbossiliciDocumento4 pagineAcidi CarbossiliciAlessandra FiorilloNessuna valutazione finora
- Appunti Chimica OrganicaDocumento53 pagineAppunti Chimica OrganicamantovanNessuna valutazione finora
- 15 - Reazioni Degli Alcheni e Degli AlchiniDocumento43 pagine15 - Reazioni Degli Alcheni e Degli AlchiniAlessandro Axl BellapiantaNessuna valutazione finora
- 5 ClassReazFrammDocumento30 pagine5 ClassReazFrammmastermv2013Nessuna valutazione finora
- Aldeidi e ChetoniDocumento21 pagineAldeidi e Chetonijuve6Nessuna valutazione finora
- ALCANI CicloalcaniDocumento15 pagineALCANI CicloalcaniCatia RiboloniNessuna valutazione finora
- Alchini PDFDocumento5 pagineAlchini PDFdavidetrtNessuna valutazione finora
- Esame Chimica Organica 1Documento6 pagineEsame Chimica Organica 1Stefano MelideoNessuna valutazione finora
- Alcani - Alcheni - Alchini - AromaticiDocumento56 pagineAlcani - Alcheni - Alchini - AromaticiМария105006Nessuna valutazione finora
- Chimica OrganicaDocumento2 pagineChimica OrganicaMargherita MaggioNessuna valutazione finora
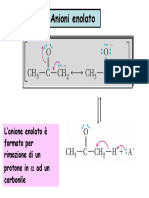
- 12) Anioni EnolatoDocumento9 pagine12) Anioni EnolatoHi HiNessuna valutazione finora
- Meccanismi Di Reazione in Chimica Organica (B.J.Kakos)Documento27 pagineMeccanismi Di Reazione in Chimica Organica (B.J.Kakos)Vassily KakosNessuna valutazione finora
- Teoria ChimicaDocumento7 pagineTeoria ChimicaWilliam ParadisoNessuna valutazione finora
- Programma Chimica Organica L Biologia Cellulare MolecolareDocumento3 pagineProgramma Chimica Organica L Biologia Cellulare MolecolareLe RodriguezNessuna valutazione finora
- Quimica AlquiloDocumento20 pagineQuimica AlquiloDelia FloresNessuna valutazione finora
- Chimica Organica - Alcani e AlcheniDocumento6 pagineChimica Organica - Alcani e AlcheniSerena GiordanoNessuna valutazione finora
- Radicali LIBERIDocumento11 pagineRadicali LIBERIilsoffionissimoNessuna valutazione finora
- CHIMICADocumento27 pagineCHIMICAgio cagliariNessuna valutazione finora
- Acilazione Di Friedel-CraftsDocumento3 pagineAcilazione Di Friedel-CraftsPaolo MelgariNessuna valutazione finora
- AMMINEDocumento40 pagineAMMINEassunta iannoneNessuna valutazione finora
- ElettrochimicaDocumento39 pagineElettrochimicaHernan MarianiNessuna valutazione finora
- Saggi Di Tollens e Di Fehling.Documento4 pagineSaggi Di Tollens e Di Fehling.Andrea Gerhard Lutz "AF"50% (2)
- Alcoli Eteri e TioliDocumento25 pagineAlcoli Eteri e TioliRenato GavaNessuna valutazione finora
- Aldeidi e ChetoniDocumento16 pagineAldeidi e ChetoniNicoleNessuna valutazione finora
- Appunti COSN 3aDocumento35 pagineAppunti COSN 3agianni fantoniNessuna valutazione finora
- Chimica OrganicaDocumento4 pagineChimica OrganicaAlessandro IsidoriNessuna valutazione finora
- Tabella Proprietà Composti OrganiciDocumento8 pagineTabella Proprietà Composti OrganiciLovelyJadeNessuna valutazione finora
- Ioni EnolatoDocumento15 pagineIoni EnolatoWolframio74Nessuna valutazione finora
- Reazioni ZuccheriDocumento5 pagineReazioni ZuccheriTommaso PisanòNessuna valutazione finora
- Atkins GlossarioDocumento33 pagineAtkins GlossarioAndrea Bucci0% (1)
- 7 Cap23 AmmineDocumento50 pagine7 Cap23 AmmineSara FaddaNessuna valutazione finora
- Alcheni e DieniDocumento5 pagineAlcheni e DieniAlessandra FiorilloNessuna valutazione finora
- ElettrochimicaDocumento37 pagineElettrochimicaDaniele CaglieroNessuna valutazione finora
- Aldeidi e ChetoniDocumento51 pagineAldeidi e ChetoniMajorana.WEBNessuna valutazione finora
- Accumulatori per impianti ad energia rinnovabileDa EverandAccumulatori per impianti ad energia rinnovabileValutazione: 2 su 5 stelle2/5 (1)
- Il Cuore-ElettrofisiologiaDocumento47 pagineIl Cuore-ElettrofisiologiaStella FilanteNessuna valutazione finora
- 18 - Sostituzioni SullAnello AromaticoDocumento47 pagine18 - Sostituzioni SullAnello AromaticoLore96Nessuna valutazione finora
- Attività Elettrica Del CuoreDocumento26 pagineAttività Elettrica Del CuoreStella FilanteNessuna valutazione finora
- Composti Di Zolfo e FosforoDocumento34 pagineComposti Di Zolfo e FosforoStella FilanteNessuna valutazione finora
- 26.esercizi Metodi 21-05Documento12 pagine26.esercizi Metodi 21-05Virginia LuzziNessuna valutazione finora
- Organic Chemistry (Ita)Documento13 pagineOrganic Chemistry (Ita)EUROPITONessuna valutazione finora
- E6 ChetoniDocumento3 pagineE6 ChetoniDiletta MontanelliNessuna valutazione finora
- Cap.14 Aldeidi e ChetoniDocumento38 pagineCap.14 Aldeidi e ChetoniGabriele GuerraNessuna valutazione finora
- Esame Scienze Di Base - Prop. BiochimicaDocumento1 paginaEsame Scienze Di Base - Prop. BiochimicaDio BeatsNessuna valutazione finora
- Esercizi OrganicaDocumento18 pagineEsercizi OrganicaBiagio CastronovoNessuna valutazione finora