Potrebbero piacerti anche
- Análisis Clínico II Determinación de HLC-CDocumento16 pagineAnálisis Clínico II Determinación de HLC-CDilcerCernaNessuna valutazione finora
- Lipoproteína VLDL, LDL Y HDL: Omar Martínez Y Felipe Morales 2 9 / 0 3 / 2 0 1 9Documento9 pagineLipoproteína VLDL, LDL Y HDL: Omar Martínez Y Felipe Morales 2 9 / 0 3 / 2 0 1 9Felipe MoralesNessuna valutazione finora
- Lipoproteínas Plasmáticas PRIMERA PARTEDocumento18 pagineLipoproteínas Plasmáticas PRIMERA PARTEWENDOLI MALDONADO HERNANDEZNessuna valutazione finora
- Taller - RESUMEN ARTICULOS LIPIDOSDocumento9 pagineTaller - RESUMEN ARTICULOS LIPIDOSjoelmangones329Nessuna valutazione finora
- Dislipidemias y ArteriosclerosisDocumento20 pagineDislipidemias y ArteriosclerosisClaudia Patricia Ballen GarzonNessuna valutazione finora
- LIPPOPROTEINASDocumento18 pagineLIPPOPROTEINASLeydi AY100% (1)
- Metabolismo de Las LipoproteinasDocumento25 pagineMetabolismo de Las LipoproteinasDennys CuevaNessuna valutazione finora
- Guia LipoproteinasDocumento8 pagineGuia LipoproteinasManuel de Jesus100% (1)
- Lipoproteínas PlasmáticasDocumento11 pagineLipoproteínas PlasmáticasF FretesNessuna valutazione finora
- Farmacologia I - Unidad IIIDocumento102 pagineFarmacologia I - Unidad IIIadrngilson11Nessuna valutazione finora
- FISIOPATOLOGÍA Dislipidemias CorregidoDocumento6 pagineFISIOPATOLOGÍA Dislipidemias CorregidoAndrea Asca La TorreNessuna valutazione finora
- Lipoproteínas y Proteína ABCA1Documento10 pagineLipoproteínas y Proteína ABCA1SergioNessuna valutazione finora
- 32 Dislipemias 2021Documento17 pagine32 Dislipemias 2021Estela EscalanteNessuna valutazione finora
- 2 Metabolismo de LípidosDocumento64 pagine2 Metabolismo de LípidosAntonella SantaellaNessuna valutazione finora
- Liproteínas y Sus Alteraciones - Documento de InvestigaciónDocumento22 pagineLiproteínas y Sus Alteraciones - Documento de InvestigaciónandreaNessuna valutazione finora
- Lipoproteínas PDFDocumento4 pagineLipoproteínas PDFMónica GonzálezNessuna valutazione finora
- T-28 Metabolismo LipoproteinasDocumento21 pagineT-28 Metabolismo LipoproteinasLeydi AYNessuna valutazione finora
- Determinación de HDL ColesterolDocumento16 pagineDeterminación de HDL ColesterolCarmen Jacobe QuichcaNessuna valutazione finora
- Guia de Estudio Lipoproteinas Plasmaticas 3-2021Documento5 pagineGuia de Estudio Lipoproteinas Plasmaticas 3-2021christian100% (1)
- Metabolismo de Lipoproteínas-1Documento10 pagineMetabolismo de Lipoproteínas-1Edher ArturoNessuna valutazione finora
- NUT - Química Metabolismo LipidosDocumento11 pagineNUT - Química Metabolismo LipidosMateo CavallerisNessuna valutazione finora
- Dislipidemias y AterogénesisDocumento21 pagineDislipidemias y AterogénesisgustavoNessuna valutazione finora
- Lipoproteinas Analisis p.3Documento12 pagineLipoproteinas Analisis p.3jkr210% (1)
- 4to Parcial BTFDocumento106 pagine4to Parcial BTFAvril Correa AparicioNessuna valutazione finora
- 2-2015.TEMA 7. Lipoproteínas. 4ta Parte PDFDocumento18 pagine2-2015.TEMA 7. Lipoproteínas. 4ta Parte PDFDaniel Salazar ArangoNessuna valutazione finora
- Taller Final Sobre Metabolismo de Lipidos y de VitaminasDocumento15 pagineTaller Final Sobre Metabolismo de Lipidos y de VitaminasSteffy Erazo BaqueroNessuna valutazione finora
- DISLIPIDEMIASDocumento17 pagineDISLIPIDEMIASVanny RuizNessuna valutazione finora
- Sem 17 Metabolismo Lipoproteinas PDFDocumento35 pagineSem 17 Metabolismo Lipoproteinas PDFAngiie PreyslerNessuna valutazione finora
- Metabolismo de LipoproteinasDocumento9 pagineMetabolismo de LipoproteinasCristhian Tarrillo EspinozaNessuna valutazione finora
- Metabolismo de LípidosDocumento14 pagineMetabolismo de LípidosNinfa LópezNessuna valutazione finora
- Modulo II DislipidemiaDocumento26 pagineModulo II Dislipidemiapalomazul007Nessuna valutazione finora
- Movilización de Lípidos. Lipoproteínas 1Documento16 pagineMovilización de Lípidos. Lipoproteínas 1Kiefer MedinaNessuna valutazione finora
- LipoproteinasDocumento26 pagineLipoproteinasLuis Gomez SanchezNessuna valutazione finora
- Super MejoradoDocumento8 pagineSuper MejoradoCorina PeralesNessuna valutazione finora
- HipercolesterolemiaDocumento9 pagineHipercolesterolemiaGus AnitaNessuna valutazione finora
- Tema 16 EndoDocumento18 pagineTema 16 EndoUniversal UniversalNessuna valutazione finora
- Lipoproteinas 1Documento7 pagineLipoproteinas 1Celeste Guadalupe Gómez RamosNessuna valutazione finora
- Mecanismo de Regulación de Las LipoproteínasDocumento3 pagineMecanismo de Regulación de Las LipoproteínasGonzalo RojasNessuna valutazione finora
- Cuestionario de Lipoproteínas y Colesterol Resuelto PDFDocumento6 pagineCuestionario de Lipoproteínas y Colesterol Resuelto PDFvale100% (1)
- Transporte de Lipidos - LipoproteinasDocumento21 pagineTransporte de Lipidos - Lipoproteinasirene AuzaNessuna valutazione finora
- 2 Parcial Corrillo 3 Metabolismo de LipoproteinasDocumento9 pagine2 Parcial Corrillo 3 Metabolismo de Lipoproteinaskaren.pina050703Nessuna valutazione finora
- 9-Bioquimica Facultad 20 de OctubreDocumento41 pagine9-Bioquimica Facultad 20 de OctubreVizcaino Rojas JosèNessuna valutazione finora
- Expocisión de LipoproteinasDocumento10 pagineExpocisión de LipoproteinasSaid AlvaradoNessuna valutazione finora
- Metabolis Mo Del Colester OLDocumento22 pagineMetabolis Mo Del Colester OLIMVASCHMNessuna valutazione finora
- Metabolismo Lipoproteinas PresentacionDocumento24 pagineMetabolismo Lipoproteinas PresentacionAnjeoly Mariel Martinez100% (3)
- 3.4 Metabolismo LipoproteinasDocumento23 pagine3.4 Metabolismo LipoproteinasNoelia LaraNessuna valutazione finora
- Lípidos Metabolismo y Enfermedades PDFDocumento12 pagineLípidos Metabolismo y Enfermedades PDFJuliet EnriquezNessuna valutazione finora
- Seminario y TP de Lipoprote NasDocumento37 pagineSeminario y TP de Lipoprote Nasnancy matielloNessuna valutazione finora
- Lipoproteinas. InformeDocumento27 pagineLipoproteinas. Informejohamydiaz100% (1)
- Guia LipoproteinasDocumento16 pagineGuia LipoproteinasJesu Herrera67% (3)
- Lipoproteinas y Hormonas EsteroidesDocumento42 pagineLipoproteinas y Hormonas EsteroidesChelsea Wisar EnriquezNessuna valutazione finora
- Los Lípidos NutricionDocumento3 pagineLos Lípidos NutricionJanna SosaNessuna valutazione finora
- Los LípidosDocumento3 pagineLos Lípidosyesenia garciaNessuna valutazione finora
- Exposición de Bioquímica KarenDocumento9 pagineExposición de Bioquímica KarenSaid AlvaradoNessuna valutazione finora
- Metabolismo LipoproteinasDocumento44 pagineMetabolismo LipoproteinasUSMP FN ARCHIVOS100% (7)
- Farmacoterapia de Las DislipidemiasDocumento8 pagineFarmacoterapia de Las DislipidemiasDiego Zapata100% (1)
- Tema 11 LipoproteinasDocumento27 pagineTema 11 LipoproteinasDiego Quispe TorresNessuna valutazione finora
- La Ciencia Del Metabolismo: Metabolismo sin rodeosDa EverandLa Ciencia Del Metabolismo: Metabolismo sin rodeosValutazione: 5 su 5 stelle5/5 (1)
- Acceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017Da EverandAcceso a Universidad para Mayores de 25 años. Biología 2013-2017.: Solucionario Pruebas 2013-2017Nessuna valutazione finora
- ANORMALIDADES VASCULAR Signos RadiológicosDocumento2 pagineANORMALIDADES VASCULAR Signos RadiológicosCarlosSonoNessuna valutazione finora
- Guion Artritis ReumatoideDocumento11 pagineGuion Artritis ReumatoideCarlosSonoNessuna valutazione finora
- Guion Lab. ClinicoDocumento9 pagineGuion Lab. ClinicoCarlosSonoNessuna valutazione finora
- Auscultacion AbdominalDocumento3 pagineAuscultacion AbdominalCarlosSonoNessuna valutazione finora
- Historia ClínicaDocumento1 paginaHistoria ClínicaCarlosSonoNessuna valutazione finora
- Correlato Fisiopatológico Del Síndrome PrincipalDocumento1 paginaCorrelato Fisiopatológico Del Síndrome PrincipalCarlosSonoNessuna valutazione finora
- Fisiopatología: Síndrome AscíticoDocumento1 paginaFisiopatología: Síndrome AscíticoCarlosSonoNessuna valutazione finora
- Histología Linfoma de HodgkinDocumento3 pagineHistología Linfoma de HodgkinCarlosSonoNessuna valutazione finora
- s8 - Radioanatomia Osea - Grupo m31 - Dr. Humberto Rosas.Documento15 pagines8 - Radioanatomia Osea - Grupo m31 - Dr. Humberto Rosas.CarlosSonoNessuna valutazione finora
- Concurso NavideñoDocumento1 paginaConcurso NavideñoCarlosSonoNessuna valutazione finora
- 2 PsicofisiologíaDocumento2 pagine2 PsicofisiologíaCarlosSonoNessuna valutazione finora
- Desarrollo Embrionario Del OjoDocumento2 pagineDesarrollo Embrionario Del OjoCarlosSonoNessuna valutazione finora
- Caso Clínico 10Documento6 pagineCaso Clínico 10CarlosSonoNessuna valutazione finora
- Experimento 2Documento3 pagineExperimento 2CarlosSonoNessuna valutazione finora
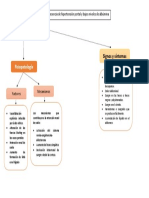
- Patología Práctica - Semana 3Documento1 paginaPatología Práctica - Semana 3CarlosSonoNessuna valutazione finora
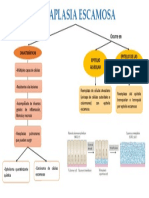
- Metaplasia EscamosaDocumento1 paginaMetaplasia EscamosaCarlosSono100% (1)
- S 3. 3. Exposición S3 Etica Clínica 2Documento5 pagineS 3. 3. Exposición S3 Etica Clínica 2CarlosSonoNessuna valutazione finora
- Bioética Marco Teórico Punto 1 y 2Documento5 pagineBioética Marco Teórico Punto 1 y 2CarlosSonoNessuna valutazione finora
- 5.-¿Cuáles Son Las Diferencias de Signos y Síntomas Entre Coma Estructural y Coma Metabólico?Documento4 pagine5.-¿Cuáles Son Las Diferencias de Signos y Síntomas Entre Coma Estructural y Coma Metabólico?CarlosSonoNessuna valutazione finora
- Preg 9 y 10Documento5 paginePreg 9 y 10CarlosSonoNessuna valutazione finora
- Prehistoria de La PatologíaDocumento4 paginePrehistoria de La PatologíaCarlosSonoNessuna valutazione finora
- Experimento 1Documento5 pagineExperimento 1CarlosSonoNessuna valutazione finora
- Punto IsoelectricoDocumento5 paginePunto IsoelectricoAntonio CruzNessuna valutazione finora
- EpigeneticaDocumento31 pagineEpigeneticac6sar6e6g6mez6r100% (2)
- Ehrlichia CanisDocumento5 pagineEhrlichia CanisNaruto ShikamaruNessuna valutazione finora
- Ratón de Bolsillo Genética - EvlyngDocumento6 pagineRatón de Bolsillo Genética - EvlyngEVELYN VIANEYNessuna valutazione finora
- EicosanoidesDocumento7 pagineEicosanoidesjonahiNessuna valutazione finora
- Ruta Embden Meyerhof PDFDocumento2 pagineRuta Embden Meyerhof PDFBrandonCaballeroNessuna valutazione finora
- BACTERIAS MilDocumento195 pagineBACTERIAS MilKaycee milenaNessuna valutazione finora
- Wuolah Free Tema 2 Las Bases de La HerenciaDocumento4 pagineWuolah Free Tema 2 Las Bases de La HerenciaIker IgleroNessuna valutazione finora
- Vitaminas C, B1 y B2Documento39 pagineVitaminas C, B1 y B2andreigustvNessuna valutazione finora
- Aminoacidos No Esenciales en Los PollosDocumento2 pagineAminoacidos No Esenciales en Los PollosGrushenka LoorNessuna valutazione finora
- Tema 2. Psicobiología, Psiconeuroinmunología y Neuroendocrinología.Documento27 pagineTema 2. Psicobiología, Psiconeuroinmunología y Neuroendocrinología.Emilio Marin Postigo DEP 16Nessuna valutazione finora
- Imed Leloir Choice Biologia Celular Catedras 2 y 3Documento16 pagineImed Leloir Choice Biologia Celular Catedras 2 y 3Jose Alberto Aguilar HuamanNessuna valutazione finora
- BIOMOLECULASDocumento3 pagineBIOMOLECULASLuisa CastañoNessuna valutazione finora
- Oxidacion Aa y UreaDocumento19 pagineOxidacion Aa y UreaAremi Gordillo StarkNessuna valutazione finora
- Taller - BLAST TerminadoDocumento9 pagineTaller - BLAST TerminadoJuan José Camacho T.Nessuna valutazione finora
- Biologia 02 Miguel Cortez OyolaDocumento3 pagineBiologia 02 Miguel Cortez Oyolacalosra50% (2)
- Fermentacion LacticaDocumento12 pagineFermentacion LacticaMario Arriola VertizNessuna valutazione finora
- Complejos Remodeladores de Cromatina - LeydiDocumento11 pagineComplejos Remodeladores de Cromatina - LeydiBryan SocorroNessuna valutazione finora
- Sistemas Energéticos en El DeporteDocumento4 pagineSistemas Energéticos en El DeporteHarvy SuárezNessuna valutazione finora
- LIPOGENESISDocumento12 pagineLIPOGENESISDanery HurtadoNessuna valutazione finora
- Solucionario Exam BioDocumento2 pagineSolucionario Exam BiosupriyaNessuna valutazione finora
- Alba Aguayo Et Al., 2013 - QPCR (Tiempo Real)Documento5 pagineAlba Aguayo Et Al., 2013 - QPCR (Tiempo Real)Tatiana Sanchez AlvarezNessuna valutazione finora
- Article Tratamiento KeratinaDocumento2 pagineArticle Tratamiento Keratinachunkywarehouse37Nessuna valutazione finora
- Informe Fago Lambda M1 LACC2Documento9 pagineInforme Fago Lambda M1 LACC2Noelia Deibe PérezNessuna valutazione finora
- Informe de TinciónDocumento18 pagineInforme de TinciónJuan Miguel Morelo AlemanNessuna valutazione finora
- Tcta4 U2 Sesión 07Documento4 pagineTcta4 U2 Sesión 07herbyn10Nessuna valutazione finora
- Mapa Conceptual y Su Significado Estilo ModernoDocumento4 pagineMapa Conceptual y Su Significado Estilo ModernoAgni Rajado VíboraNessuna valutazione finora
- MutacionesDocumento48 pagineMutacioneshenryNessuna valutazione finora
- Ácido FólicoDocumento34 pagineÁcido FólicoChristian JaraNessuna valutazione finora
- Pract 6. Actividad EnzimáticaDocumento8 paginePract 6. Actividad EnzimáticaFischer Sánchez LeivaNessuna valutazione finora