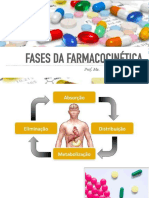
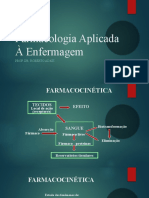
Potrebbero piacerti anche
- Aula 3Documento20 pagineAula 3beatriizzoliveira13Nessuna valutazione finora
- Resumo Farmacologia - Pag 09 e 10Documento5 pagineResumo Farmacologia - Pag 09 e 10Lucas OliveiraNessuna valutazione finora
- Aula 1 - Princípios de Farmacocinética (Atualização)Documento7 pagineAula 1 - Princípios de Farmacocinética (Atualização)Lázaro Victor Santos MendonçaNessuna valutazione finora
- Farmacologia (Cinética) - NandaDocumento40 pagineFarmacologia (Cinética) - NandaGuilherme Sen Bressani0% (1)
- Aula 03Documento37 pagineAula 03karina kelly100% (1)
- 2 - FarmacocinéticaDocumento15 pagine2 - FarmacocinéticaGabriele BassoNessuna valutazione finora
- Sessão 1 - Grupo OnlineDocumento34 pagineSessão 1 - Grupo OnlineNelito SangulaNessuna valutazione finora
- Farmacocinética - Farmacologia Aplicada À EnfermagemDocumento23 pagineFarmacocinética - Farmacologia Aplicada À EnfermagemWagner CostaNessuna valutazione finora
- Resumo FarmacologiaDocumento17 pagineResumo FarmacologiaJoão Vitor Fernandes da CunhaNessuna valutazione finora
- Psicofarmaco 3Documento46 paginePsicofarmaco 3Thais EmunahNessuna valutazione finora
- Apostila - FarmacologiaDocumento21 pagineApostila - Farmacologiacamillecramos100% (2)
- Apostila de Farmacocinetica - Adreanne OliveiraDocumento15 pagineApostila de Farmacocinetica - Adreanne OliveiraAdreanne OliveiraNessuna valutazione finora
- Farmacologia para Nutrição - FarmacocinéticaDocumento74 pagineFarmacologia para Nutrição - FarmacocinéticaMalu Novais100% (1)
- AULA 04 - Psicofarmacologia - UniFioDocumento46 pagineAULA 04 - Psicofarmacologia - UniFioduarteferreirafabianaNessuna valutazione finora
- Processos FarmacocinéticosDocumento78 pagineProcessos Farmacocinéticosmirellyarcanjo6Nessuna valutazione finora
- Absorção e Distribuição de FármacosDocumento58 pagineAbsorção e Distribuição de FármacosSandy Lima100% (1)
- FarmacocinéticaDocumento11 pagineFarmacocinéticaAnonymous Lc6BcBTQNessuna valutazione finora
- Estudo Dirigido Farmacologia RevisãoDocumento7 pagineEstudo Dirigido Farmacologia Revisãocarlosjuniorr170Nessuna valutazione finora
- 01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICADocumento55 pagine01 Aula 03 Farmacocinetica - FARMACOLOGIA BÁSICAandre almeida100% (1)
- 3 - Biotransformação e Excreção de FármacosDocumento37 pagine3 - Biotransformação e Excreção de FármacosWendell OliveiraNessuna valutazione finora
- Vias de Administração de FármacosDocumento16 pagineVias de Administração de FármacosMiguel Borges CândidoNessuna valutazione finora
- FarmacocinéticaDocumento60 pagineFarmacocinéticaednsinfNessuna valutazione finora
- Processos FarmacocinéticosDocumento61 pagineProcessos FarmacocinéticosSabrina FerreiraNessuna valutazione finora
- Absorção, Distribuição e LigaçãoDocumento63 pagineAbsorção, Distribuição e LigaçãoMariana Assis Paniago100% (1)
- Ficha para AlunosDocumento11 pagineFicha para AlunosDanilo RaulNessuna valutazione finora
- Farmacocinética PDFDocumento47 pagineFarmacocinética PDFSuely Ramlow Robson CanalNessuna valutazione finora
- Farmacologia Aplicada À NutriçãoDocumento6 pagineFarmacologia Aplicada À Nutriçãolaranutri2019.1Nessuna valutazione finora
- FarmacocineticaDocumento5 pagineFarmacocineticaRodolfo MarquesNessuna valutazione finora
- Aula 2 - FarmacocinéticaDocumento12 pagineAula 2 - Farmacocinéticanego gato100% (1)
- Farmacocinética - Absorção, Distribuição, Biotransformação e EliminaçãoDocumento5 pagineFarmacocinética - Absorção, Distribuição, Biotransformação e Eliminaçãomarcelo_paoli100% (1)
- Questionario FarmacologiaDocumento14 pagineQuestionario FarmacologiaCristiane FernandesNessuna valutazione finora
- FARMACOLOGIADocumento6 pagineFARMACOLOGIABruna KelletNessuna valutazione finora
- Farmacocinética - Absorção e DistribuiçãoDocumento12 pagineFarmacocinética - Absorção e Distribuiçãoauriculoterapia.wesNessuna valutazione finora
- Farmacocinética e Vias de AdministraçãoDocumento44 pagineFarmacocinética e Vias de AdministraçãoGabriela MirandaNessuna valutazione finora
- Princípios Básicos de FarmacologiaDocumento8 paginePrincípios Básicos de Farmacologiajorge eryNessuna valutazione finora
- Roteiro ADMEDocumento5 pagineRoteiro ADMENayara BragaNessuna valutazione finora
- Princípios FarmacocinéticosDocumento43 paginePrincípios FarmacocinéticosSilvania MartinsNessuna valutazione finora
- Fcinética - Parte 1Documento35 pagineFcinética - Parte 1Maryana LetíciaNessuna valutazione finora
- Aps Farmaco I - V1 - 2020 - Farmaco DinâmicaDocumento9 pagineAps Farmaco I - V1 - 2020 - Farmaco DinâmicaRodolfo Ribeiro FerreiraNessuna valutazione finora
- Resumo Aula 1, 2 e 3Documento27 pagineResumo Aula 1, 2 e 3Caio AmaralNessuna valutazione finora
- FarmacocinéticaDocumento6 pagineFarmacocinéticapaty201645Nessuna valutazione finora
- Farmacologia Clinica CAFARDocumento13 pagineFarmacologia Clinica CAFARkarol souzaNessuna valutazione finora
- Farmacocinética PDFDocumento60 pagineFarmacocinética PDFTimoteo VicenteNessuna valutazione finora
- Documento 2Documento4 pagineDocumento 2Carine Soares BrolloNessuna valutazione finora
- 8 - Depuração Do FármacoDocumento25 pagine8 - Depuração Do FármacoSamba Antonio IntumbaNessuna valutazione finora
- FarmacologiaDocumento13 pagineFarmacologiajean paulinoNessuna valutazione finora
- FarmacociDocumento51 pagineFarmacociSantiago Vital Freitas100% (2)
- RelatórioDocumento12 pagineRelatóriokemillysouza18Nessuna valutazione finora
- Resumo - Natália BonfáDocumento66 pagineResumo - Natália BonfáCaio AmaralNessuna valutazione finora
- Aulao Cinetica e Dinamica Final PDFDocumento126 pagineAulao Cinetica e Dinamica Final PDFTiago De Braga SoaresNessuna valutazione finora
- Resumo para Prova de FarmacologiaDocumento35 pagineResumo para Prova de FarmacologiaDébora MartinsNessuna valutazione finora
- Aulas 2 e 3 FarmacocinéticaDocumento77 pagineAulas 2 e 3 FarmacocinéticaFabio Ramalheiro100% (1)
- Farmacologia - Resumo Do GoodmanDocumento35 pagineFarmacologia - Resumo Do Goodmanyurimusso100% (3)
- 2023 Aula Teórica de FarmacocinéticaDocumento70 pagine2023 Aula Teórica de FarmacocinéticaGiovana BragaNessuna valutazione finora
- FARMACOCINETICADocumento7 pagineFARMACOCINETICAh.sanzNessuna valutazione finora
- Tutoria 2Documento8 pagineTutoria 2Rafael MachadoNessuna valutazione finora
- Resumo Farmacologia FARMACOCINTICADocumento6 pagineResumo Farmacologia FARMACOCINTICAFrancisco MachadoNessuna valutazione finora
- Solicitação e Interpretação de Exames Laboratoriais: Uma visão fundamentada e atualizada sobre a solicitação, interpretação e associação de alterações bioquímicas com o estado nutricional e fisiológico do paciente.Da EverandSolicitação e Interpretação de Exames Laboratoriais: Uma visão fundamentada e atualizada sobre a solicitação, interpretação e associação de alterações bioquímicas com o estado nutricional e fisiológico do paciente.Valutazione: 2 su 5 stelle2/5 (1)
- Antidepressivos, Ansiolíticos e Hipinóticos - ResumoDocumento3 pagineAntidepressivos, Ansiolíticos e Hipinóticos - ResumoAna Carla FaizNessuna valutazione finora
- Antidepressivos, Ansiolíticos e Hipinóticos - Slides PDFDocumento128 pagineAntidepressivos, Ansiolíticos e Hipinóticos - Slides PDFAna Carla Faiz100% (1)
- Água - SlidesDocumento46 pagineÁgua - SlidesAna Carla FaizNessuna valutazione finora
- Absorção e Distribuição de Fármacos - SlidesDocumento62 pagineAbsorção e Distribuição de Fármacos - SlidesAna Carla FaizNessuna valutazione finora
- Referências BibliográficasDocumento4 pagineReferências BibliográficasRoberto SantosNessuna valutazione finora
- Noções BásicasDocumento18 pagineNoções BásicasMariana Fraga de FigueiredoNessuna valutazione finora
- Livro Texto Unidade IDocumento217 pagineLivro Texto Unidade IPaola FedattoNessuna valutazione finora
- Projeto GestantesDocumento11 pagineProjeto GestantesCras BelmonteNessuna valutazione finora
- Propedêutica e Processo de Cuidar Da Saúde Do Adulto Questionário Online - Revisão Da Tentativa 02Documento4 paginePropedêutica e Processo de Cuidar Da Saúde Do Adulto Questionário Online - Revisão Da Tentativa 02bemawe4787Nessuna valutazione finora
- Desenvolvimento Oclusao Part I PDFDocumento8 pagineDesenvolvimento Oclusao Part I PDFTainá PereiraNessuna valutazione finora
- Investigação de Surto - Relatório de Investigação de Casos de Infecção Por MCR 1998 A 2009Documento52 pagineInvestigação de Surto - Relatório de Investigação de Casos de Infecção Por MCR 1998 A 2009Suzie Marie GomesNessuna valutazione finora
- Ebook Como Lidar Com A Ansiedade para EmagrecerDocumento7 pagineEbook Como Lidar Com A Ansiedade para EmagrecerRízia SouzaNessuna valutazione finora
- Roteiro para AnamneseDocumento5 pagineRoteiro para AnamneseHuddynNessuna valutazione finora
- Parecer CATEC CanforaDocumento1 paginaParecer CATEC CanforaEliliana LaurindoNessuna valutazione finora
- EJA Livro Médio - RótulosDocumento6 pagineEJA Livro Médio - RótulosLaura LopesNessuna valutazione finora
- BT DertexDocumento2 pagineBT DertexJeffersonNessuna valutazione finora
- Prova III - GabaritoDocumento1 paginaProva III - GabaritoDaniel CostaNessuna valutazione finora
- Material Educativo - Insônia CORRIGIDODocumento2 pagineMaterial Educativo - Insônia CORRIGIDOBruno LisboaNessuna valutazione finora
- Manual de Boas Práticas - Prefeitura Da Estância de Atibaia - Julho 2018Documento42 pagineManual de Boas Práticas - Prefeitura Da Estância de Atibaia - Julho 2018Fernando LuizNessuna valutazione finora
- Apostila Jaques Lacan SBPI PDFDocumento111 pagineApostila Jaques Lacan SBPI PDFRenata Tirolla100% (1)
- Propedêutica Da PeleDocumento7 paginePropedêutica Da PeleAlessandra Gonsalves AndradeNessuna valutazione finora
- Colo - MuvinlaxDocumento1 paginaColo - MuvinlaxEricson BenicioNessuna valutazione finora
- Redação Nota Mil No Enem - 10 Exemplos, Só Textos Aprovadas Pelo MEC 2Documento5 pagineRedação Nota Mil No Enem - 10 Exemplos, Só Textos Aprovadas Pelo MEC 2caio burgoNessuna valutazione finora
- Plano de Aula Do EJA 2 ADocumento7 paginePlano de Aula Do EJA 2 AArianeNessuna valutazione finora
- Cidade Amiga Animais 2 Edicao 2020Documento45 pagineCidade Amiga Animais 2 Edicao 2020Marcia OliveiraNessuna valutazione finora
- Reflexo Inato e Adq - 2Documento27 pagineReflexo Inato e Adq - 2RODRIGO100% (1)
- Gabarito SenaiDocumento3 pagineGabarito SenaiDaniela GomesNessuna valutazione finora
- Trabalho EnofiliaDocumento5 pagineTrabalho EnofiliaEssashikaNessuna valutazione finora
- Atividade Extra VirusDocumento6 pagineAtividade Extra VirusluizcarlosnovoNessuna valutazione finora
- Simulado 1 - Ciencias Da NaturezaDocumento3 pagineSimulado 1 - Ciencias Da NaturezaJose SantosNessuna valutazione finora
- Ortopedia e TraumatologiaDocumento22 pagineOrtopedia e TraumatologiaLarissa G. O'FarrilNessuna valutazione finora
- Agente Fiscal PDFDocumento9 pagineAgente Fiscal PDFVictor GonçalvesNessuna valutazione finora
- Cms Files 136797 1603806152ebook Oral UNic 2Documento22 pagineCms Files 136797 1603806152ebook Oral UNic 2Di Produções ...Nessuna valutazione finora
- Modelo DSMAVDocumento1 paginaModelo DSMAVEymard De Meira BredaNessuna valutazione finora