Potrebbero piacerti anche
- Michael G. Gore - Spectrophotometry and Spectrofluorimetry - A Practical Approach-Oxford University Press, USA (2000)Documento387 pagineMichael G. Gore - Spectrophotometry and Spectrofluorimetry - A Practical Approach-Oxford University Press, USA (2000)محمود طارق100% (2)
- Vacuum Engineering Calculations, Formulas, and Solved ExercisesDa EverandVacuum Engineering Calculations, Formulas, and Solved ExercisesValutazione: 4.5 su 5 stelle4.5/5 (2)
- An Introduction to Polymer Chemistry: The Commonwealth and International Library: Intermediate Chemistry DivisionDa EverandAn Introduction to Polymer Chemistry: The Commonwealth and International Library: Intermediate Chemistry DivisionValutazione: 3 su 5 stelle3/5 (3)
- Minimise Amine Losses in Gas and Liquid TreatingDocumento10 pagineMinimise Amine Losses in Gas and Liquid Treatings k kumar100% (2)
- L12 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocumento25 pagineL12 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyNessuna valutazione finora
- Catalyst Characterization - W6Documento33 pagineCatalyst Characterization - W6Safitri WulansariNessuna valutazione finora
- Porozni MaterijaliDocumento67 paginePorozni MaterijaliAna RisticNessuna valutazione finora
- Chapter 3-3Documento7 pagineChapter 3-3ImanNessuna valutazione finora
- L14 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocumento50 pagineL14 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyNessuna valutazione finora
- File 1 - Reactor Analysis Unit 5 FullDocumento73 pagineFile 1 - Reactor Analysis Unit 5 FullGaming is in my DNANessuna valutazione finora
- CHE S402 Chapter 4 Solid Catalysts Part3Documento8 pagineCHE S402 Chapter 4 Solid Catalysts Part3Rashmi SahooNessuna valutazione finora
- Solid CatalystsDocumento8 pagineSolid CatalystsVISHAL SHARMANessuna valutazione finora
- Chemical Reaction Engineering-II (2170501) : Semester - VII (CHEM) Chapter Name: Catalysis (Chapter 5)Documento11 pagineChemical Reaction Engineering-II (2170501) : Semester - VII (CHEM) Chapter Name: Catalysis (Chapter 5)khushbu100% (2)
- Bet-Analysis: by Brunauer-Emmett-Teller 1938Documento13 pagineBet-Analysis: by Brunauer-Emmett-Teller 1938harshit tiwariNessuna valutazione finora
- Surface Area and Porosity-1Documento37 pagineSurface Area and Porosity-1Ghana Cintai DiaNessuna valutazione finora
- Syahrul SSKM 3-Surface CharacterizationDocumento41 pagineSyahrul SSKM 3-Surface CharacterizationHusna KamiliaNessuna valutazione finora
- Bet TheoryDocumento19 pagineBet Theoryprakashom018Nessuna valutazione finora
- Thin Film Deposition - Thermal and E-Bean EvaporationDocumento96 pagineThin Film Deposition - Thermal and E-Bean EvaporationAnurag KumarNessuna valutazione finora
- Class 03Documento8 pagineClass 03Lâm LêNessuna valutazione finora
- Chapter 9 Thin Film Deposition - IIDocumento35 pagineChapter 9 Thin Film Deposition - IIFahmi Alfian SyahNessuna valutazione finora
- Introduction To Catalysis Cp2Documento35 pagineIntroduction To Catalysis Cp2Godfrey Eric MuendoNessuna valutazione finora
- Chuong 2. BE MAT TIEP XUC-PHAN TICH HOA LYDocumento50 pagineChuong 2. BE MAT TIEP XUC-PHAN TICH HOA LYntnphung.sdh222Nessuna valutazione finora
- FulltextDocumento90 pagineFulltextjokonudiNessuna valutazione finora
- Nitrogen Adsorption Isotherms On Carbonaceous Materials - Gomez-Serrano Et Al 2000Documento6 pagineNitrogen Adsorption Isotherms On Carbonaceous Materials - Gomez-Serrano Et Al 2000ianphilanderNessuna valutazione finora
- Lecturs-5 6 (Porosity and Permeability From Core PDFDocumento47 pagineLecturs-5 6 (Porosity and Permeability From Core PDFMuntasir MokhtarNessuna valutazione finora
- Lec 4Documento5 pagineLec 4Ghazy alshyalNessuna valutazione finora
- L15 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocumento15 pagineL15 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyNessuna valutazione finora
- Surface Chemistry L2Documento27 pagineSurface Chemistry L2Ilham Faturachman0% (1)
- Basic Gas ChromatographyDocumento42 pagineBasic Gas ChromatographyNofrizalNessuna valutazione finora
- Particle Size Analysis & Bet Adsorption: Presented by Joshwa JojiDocumento13 pagineParticle Size Analysis & Bet Adsorption: Presented by Joshwa JojiJoshwa JojiNessuna valutazione finora
- CH-440 NanotechnologyDocumento22 pagineCH-440 NanotechnologyAndrew SionNessuna valutazione finora
- Air Pollution: Classification of Air PollutantsDocumento33 pagineAir Pollution: Classification of Air PollutantsTanzeela RiazNessuna valutazione finora
- L16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiDocumento25 pagineL16 CRE II Heterogeneous Catalysis: Prof. K.K.Pant Department of Chemical Engineering IIT DelhiMehul VarshneyNessuna valutazione finora
- Adsorption Lecture Notes: ENVE542 Air Pollution Control Technologies by Dr. Pınar Ergenekon GYTE 2010 FallDocumento51 pagineAdsorption Lecture Notes: ENVE542 Air Pollution Control Technologies by Dr. Pınar Ergenekon GYTE 2010 Fallimran24Nessuna valutazione finora
- Adsorption January 2023Documento49 pagineAdsorption January 2023Saria ChowdhuryNessuna valutazione finora
- Catalyst Characterization PPT NewDocumento34 pagineCatalyst Characterization PPT NewTesfaye KassawNessuna valutazione finora
- Removal of Volatile Organic Compound by Activated Carbon FiberDocumento14 pagineRemoval of Volatile Organic Compound by Activated Carbon FiberArash AbbasiNessuna valutazione finora
- Research On Influence Factors On Determination of Specific Surface Area of Carbon Materical by N2 Adsorption MethodDocumento6 pagineResearch On Influence Factors On Determination of Specific Surface Area of Carbon Materical by N2 Adsorption MethodTyDolla ChicoNessuna valutazione finora
- Dokumen - Tips Reservoir-PetrophysicsDocumento42 pagineDokumen - Tips Reservoir-PetrophysicsinggitalfthhNessuna valutazione finora
- Heterogeneous Catalysis: Most Heterogeneous Catalysts Are Supported CatalystsDocumento30 pagineHeterogeneous Catalysis: Most Heterogeneous Catalysts Are Supported CatalystsAhom CKNessuna valutazione finora
- Drag CoefficientDocumento61 pagineDrag CoefficientSaurabh SharmaNessuna valutazione finora
- ADSORPSI II. 31 MAr 21Documento15 pagineADSORPSI II. 31 MAr 21YumaNurAlfathNessuna valutazione finora
- Laboratory Report 3Documento3 pagineLaboratory Report 3api-3864822Nessuna valutazione finora
- Micp PDFDocumento31 pagineMicp PDFAngela rismaNessuna valutazione finora
- Adsorption Isotherm HLDocumento26 pagineAdsorption Isotherm HLDhea RahmayantiNessuna valutazione finora
- C.R.E. - II (All Units)Documento88 pagineC.R.E. - II (All Units)Mohamed Shahid100% (1)
- Physical Chemistry 2 - Surface Phenomena and AdsorptionDocumento43 paginePhysical Chemistry 2 - Surface Phenomena and AdsorptionNguyễn Thu HàNessuna valutazione finora
- Seminar JSD - IIDocumento102 pagineSeminar JSD - IIleizar_death64Nessuna valutazione finora
- 3-Adsorption EquilibriaDocumento33 pagine3-Adsorption EquilibriaImran KhanNessuna valutazione finora
- Quadrupole and Ion Trap Mass Analysers and An Introduction To ResolutionDocumento31 pagineQuadrupole and Ion Trap Mass Analysers and An Introduction To ResolutionOnur UyanıkNessuna valutazione finora
- 12월 02일 AC 강의Documento22 pagine12월 02일 AC 강의DooilNessuna valutazione finora
- Lec 9Documento14 pagineLec 9ankitsharma67280Nessuna valutazione finora
- Density LogDocumento24 pagineDensity LogBarqun DzulqurnainNessuna valutazione finora
- Adsorption ReviewDocumento29 pagineAdsorption ReviewJason PacilNessuna valutazione finora
- 3 Thin Film Deposition - IIDocumento28 pagine3 Thin Film Deposition - IIPhụng NguyễnNessuna valutazione finora
- 05 Characterization of Heterogeneous Catalyst (256512)Documento86 pagine05 Characterization of Heterogeneous Catalyst (256512)kitchaya UHVNessuna valutazione finora
- --Documento70 pagine--The TinyStrikerNessuna valutazione finora
- Influence of Chemical Reaction Rate, Diffusion and Pore Structure On The Regeneration of A Coked Al O - CatalystDocumento13 pagineInfluence of Chemical Reaction Rate, Diffusion and Pore Structure On The Regeneration of A Coked Al O - CatalystArif HidayatNessuna valutazione finora
- Accepted Manuscript: Chemical Engineering JournalDocumento41 pagineAccepted Manuscript: Chemical Engineering Journalmohsen ranjbarNessuna valutazione finora
- 03 Catalyst CharacterizationDocumento39 pagine03 Catalyst CharacterizationMegan TorresNessuna valutazione finora
- Fundamentals of Fluidized-Bed Chemical Processes: Butterworths Monographs in Chemical EngineeringDa EverandFundamentals of Fluidized-Bed Chemical Processes: Butterworths Monographs in Chemical EngineeringNessuna valutazione finora
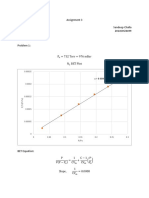
- Assignment 3Documento4 pagineAssignment 3Sandeep ChallaNessuna valutazione finora
- Interpretation:: CH NH BFDocumento2 pagineInterpretation:: CH NH BFSandeep ChallaNessuna valutazione finora
- L26 27 PDFDocumento35 pagineL26 27 PDFSandeep ChallaNessuna valutazione finora
- Interpretation:: Lewis Base Lewis AcidDocumento4 pagineInterpretation:: Lewis Base Lewis AcidSandeep ChallaNessuna valutazione finora
- 3 2PPDocumento3 pagine3 2PPSandeep ChallaNessuna valutazione finora
- Assignment 2 3Documento3 pagineAssignment 2 3Sandeep Challa0% (1)
- 3 2PPDocumento3 pagine3 2PPSandeep ChallaNessuna valutazione finora
- Distillation Column DesignDocumento20 pagineDistillation Column DesignSandeep Challa100% (1)
- COMMWIP Assignment - Hemant VermaDocumento3 pagineCOMMWIP Assignment - Hemant VermaSandeep ChallaNessuna valutazione finora
- B.tech Chemical DatabaseDocumento5 pagineB.tech Chemical DatabaseSandeep ChallaNessuna valutazione finora
- OrientDocumento20 pagineOrientSandeep ChallaNessuna valutazione finora
- DHDS ProcessDocumento9 pagineDHDS ProcessSandeep ChallaNessuna valutazione finora
- Activity No. 3 Proteins SasDocumento2 pagineActivity No. 3 Proteins SasHecStoryNessuna valutazione finora
- Mercury Contamination RisksDocumento78 pagineMercury Contamination RisksguruhnurizalNessuna valutazione finora
- Iso 12952 6 2021Documento38 pagineIso 12952 6 2021Jim FrenkenNessuna valutazione finora
- 8485 PDFDocumento1 pagina8485 PDFtakudzwaNessuna valutazione finora
- Applications of Equations of StateDocumento17 pagineApplications of Equations of StateJatin RamboNessuna valutazione finora
- For Chemistry Gas Laws Handout 1 2Documento2 pagineFor Chemistry Gas Laws Handout 1 2aira sharidaNessuna valutazione finora
- 219-Article Text-1649-1-10-20230601Documento14 pagine219-Article Text-1649-1-10-20230601mahdum afdhaNessuna valutazione finora
- Activity 10: Standard Solutions - Weighing Method: Try This: To Prepare 250 CMDocumento9 pagineActivity 10: Standard Solutions - Weighing Method: Try This: To Prepare 250 CMThilagavathyNessuna valutazione finora
- Science PapersDocumento116 pagineScience Papersstar007865Nessuna valutazione finora
- Cambridge IGCSE: Combined Science 0653/61Documento16 pagineCambridge IGCSE: Combined Science 0653/61Hin Wa LeungNessuna valutazione finora
- Portal MethodDocumento16 paginePortal MethodRajendra prasad G.CNessuna valutazione finora
- Adsorption of Methylene Blue Dye From Aqueous Solution by Nano Nickle Oxide CatalystDocumento31 pagineAdsorption of Methylene Blue Dye From Aqueous Solution by Nano Nickle Oxide Catalyst4 Aya AshrafNessuna valutazione finora
- Daffodil International University: Textile Testing & Quality Control-3Documento11 pagineDaffodil International University: Textile Testing & Quality Control-3Dyeing DyeingNessuna valutazione finora
- GCE Singapore-Cambridge Pure Chemistry NotesDocumento21 pagineGCE Singapore-Cambridge Pure Chemistry NotesChong56Nessuna valutazione finora
- Journal of Environmental Chemical Engineering: SciencedirectDocumento8 pagineJournal of Environmental Chemical Engineering: Sciencedirectcuc12cptNessuna valutazione finora
- USP-NF Acepromazine Maleate InjectionDocumento2 pagineUSP-NF Acepromazine Maleate InjectionStalin VacaNessuna valutazione finora
- Science - Test Paper - X - CH 5Documento4 pagineScience - Test Paper - X - CH 5Víshál RánáNessuna valutazione finora
- BaronDocumento54 pagineBaronBARON NKETANINessuna valutazione finora
- Pulleys For Flat Belts Idlers For Flat BeltsDocumento1 paginaPulleys For Flat Belts Idlers For Flat BeltsShania SNessuna valutazione finora
- Exxonmobil™ Hdpe HD 7960.13: High Density Polyethylene ResinDocumento2 pagineExxonmobil™ Hdpe HD 7960.13: High Density Polyethylene ResindianaNessuna valutazione finora
- Surface Roughness Produced by Different Manufacturing Process PDFDocumento4 pagineSurface Roughness Produced by Different Manufacturing Process PDFfef123123Nessuna valutazione finora
- Galv Certificate New PDFDocumento1 paginaGalv Certificate New PDFQc QatarNessuna valutazione finora
- Solids Analysis PDFDocumento2 pagineSolids Analysis PDFEshwar NukalaNessuna valutazione finora
- Publications Récentes:: June (2020) - 3-H. Hammania, M. El Achabyd, K. El Harfi, M. A. El Mhammedi andDocumento2 paginePublications Récentes:: June (2020) - 3-H. Hammania, M. El Achabyd, K. El Harfi, M. A. El Mhammedi andkhalifa El harfiNessuna valutazione finora
- Experiment 3 CHM510Documento6 pagineExperiment 3 CHM510Dang HumairahNessuna valutazione finora
- High Performance Liquid Chromatography - HPLCDocumento9 pagineHigh Performance Liquid Chromatography - HPLCNirmal SharmaNessuna valutazione finora
- SekaninaiteDocumento4 pagineSekaninaiteJorgeBarredaNessuna valutazione finora
- Tonne of Refrigeration, How To Calculate Required TR - Pharma EngineeringDocumento12 pagineTonne of Refrigeration, How To Calculate Required TR - Pharma EngineeringpratikNessuna valutazione finora