Potrebbero piacerti anche
- Effects of Liquefaction On StructuresDocumento38 pagineEffects of Liquefaction On Structurestusharghosh86% (14)
- Lecture Notes Chapter 4 of Evans PDEDocumento7 pagineLecture Notes Chapter 4 of Evans PDEJoey WachtveitlNessuna valutazione finora
- Beam Column ConnectionDocumento2 pagineBeam Column ConnectionHAZIRACFS SURATNessuna valutazione finora
- Calculating The System Head PDFDocumento9 pagineCalculating The System Head PDFIrfanshah2013Nessuna valutazione finora
- Rectangular DuctDocumento67 pagineRectangular DuctAUCE9802100% (3)
- Terzaghi Bearing CapacityDocumento42 pagineTerzaghi Bearing CapacityJaireNessuna valutazione finora
- Flow Through Packed Fixed and Fluidized Beds PDFDocumento7 pagineFlow Through Packed Fixed and Fluidized Beds PDFelisya diantyNessuna valutazione finora
- Fluid Mechanics of Car EnginesDocumento3 pagineFluid Mechanics of Car EnginesRaaidhAhmedNessuna valutazione finora
- Goulds Residential Pump Guide PDFDocumento44 pagineGoulds Residential Pump Guide PDFSyed Muztuza Ali100% (1)
- Efficiency Benefits of High Performance Structured Packings: Kevin Bennett, Sulzer Chemtech Mark Pilling, Sulzer ChemtechDocumento31 pagineEfficiency Benefits of High Performance Structured Packings: Kevin Bennett, Sulzer Chemtech Mark Pilling, Sulzer Chemtechmisscolgate100% (1)
- CMMS Template - Consolidated HVACDocumento2.323 pagineCMMS Template - Consolidated HVACSundar DAACNessuna valutazione finora
- Flow and Level Measurement HandbookDocumento100 pagineFlow and Level Measurement Handbookmtayyab_786Nessuna valutazione finora
- Physics - TDS EquationsDocumento4 paginePhysics - TDS EquationsAmolNessuna valutazione finora
- 5.60 Thermodynamics & Kinetics: Mit OpencoursewareDocumento8 pagine5.60 Thermodynamics & Kinetics: Mit OpencoursewarecaptainhassNessuna valutazione finora
- The Terrible Beauty of ThermodynamicsDocumento9 pagineThe Terrible Beauty of Thermodynamicsels243Nessuna valutazione finora
- The Key Concept of Ensembles Is Based On 3 Types of SystemsDocumento3 pagineThe Key Concept of Ensembles Is Based On 3 Types of SystemsYasir joyiaNessuna valutazione finora
- School of Physics and Astronomy: Equilibrium and The Thermodynamic PotentialsDocumento4 pagineSchool of Physics and Astronomy: Equilibrium and The Thermodynamic PotentialsItalo YuriNessuna valutazione finora
- Thermo CheatDocumento24 pagineThermo Cheatali_b1367Nessuna valutazione finora
- Weatherwax Fermi Solution Manual PDFDocumento46 pagineWeatherwax Fermi Solution Manual PDFMaria Ileana LeónNessuna valutazione finora
- Thermodynamic PotentialsDocumento9 pagineThermodynamic Potentialshariz syazwanNessuna valutazione finora
- Section 2D. State Functions and Exact DifferentialsDocumento4 pagineSection 2D. State Functions and Exact DifferentialsAkib ImtihanNessuna valutazione finora
- 3A5 Examples Paper 2Documento10 pagine3A5 Examples Paper 2Pablo PeñasNessuna valutazione finora
- 0.1 Minimum Principles and Thermodynamic Potentials: F I F IDocumento15 pagine0.1 Minimum Principles and Thermodynamic Potentials: F I F Irq22222Nessuna valutazione finora
- School of Physics and Astronomy: Equilibrium and The Thermodynamic PotentialsDocumento4 pagineSchool of Physics and Astronomy: Equilibrium and The Thermodynamic PotentialsVaibhav PachauleeNessuna valutazione finora
- Che1003: Process Engineering ThermodynamicsDocumento50 pagineChe1003: Process Engineering ThermodynamicsAABID SHAIKNessuna valutazione finora
- Introduction To General Relativity Solutions 6-10Documento5 pagineIntroduction To General Relativity Solutions 6-10daveNessuna valutazione finora
- 2-Fundamental Property RelationsDocumento24 pagine2-Fundamental Property RelationsIkNessuna valutazione finora
- Thermodynamics II: 1 The First LawDocumento19 pagineThermodynamics II: 1 The First LawRandomNessuna valutazione finora
- SKF3013 Lecture 2Documento22 pagineSKF3013 Lecture 2Ain SufizaNessuna valutazione finora
- Test 3 Solution 2008Documento2 pagineTest 3 Solution 2008Manishaa Varatha RajuNessuna valutazione finora
- The Maxwell Relations: CHEM 331 Physical Chemistry Fall 2017Documento11 pagineThe Maxwell Relations: CHEM 331 Physical Chemistry Fall 2017AhmedovicMichaelNessuna valutazione finora
- First Law of Thermodyamics: U Q W U Q W Entropy Enthalpy S H U PVDocumento8 pagineFirst Law of Thermodyamics: U Q W U Q W Entropy Enthalpy S H U PVvishnu chaudharyNessuna valutazione finora
- Chem 340 - Notes 7Documento7 pagineChem 340 - Notes 7almeidaciscoNessuna valutazione finora
- Lecture 8 - Thermodynamic Potentials, Gibbs Free Energy, Etc-1Documento61 pagineLecture 8 - Thermodynamic Potentials, Gibbs Free Energy, Etc-1BENNessuna valutazione finora
- CHE 415 Module2-3Documento64 pagineCHE 415 Module2-3Osan ThorpeNessuna valutazione finora
- Thermodynamic Properties PDFDocumento18 pagineThermodynamic Properties PDFUdayan Panda100% (1)
- The Third Law of ThermodynamicDocumento7 pagineThe Third Law of ThermodynamicNurshuhada NordinNessuna valutazione finora
- 2016 - Bourquard - CAPS Technical Note On Acoustic MeasurementsDocumento26 pagine2016 - Bourquard - CAPS Technical Note On Acoustic MeasurementslakaviyNessuna valutazione finora
- Termo Fermi SolDocumento46 pagineTermo Fermi SolȘtefan RăzvanNessuna valutazione finora
- The Wave Equation of Sound: Bernd Porr October 6, 2005Documento7 pagineThe Wave Equation of Sound: Bernd Porr October 6, 2005Mudit Goel100% (1)
- Thermo PotentialDocumento16 pagineThermo PotentialSatya SinduriNessuna valutazione finora
- Duhamel's PrincipleDocumento6 pagineDuhamel's PrinciplezelihahaNessuna valutazione finora
- Losses in Fuel CellsDocumento31 pagineLosses in Fuel CellsLjubodrag SamardzicNessuna valutazione finora
- Chapter 6: Thermodynamic Properties of Real Fluids The Four Fundamental Property RelationsDocumento51 pagineChapter 6: Thermodynamic Properties of Real Fluids The Four Fundamental Property RelationsBilal AhmedNessuna valutazione finora
- Esci341 Lesson11 Energy Minimum PrincipleDocumento8 pagineEsci341 Lesson11 Energy Minimum PrincipleDilaraa AtılganNessuna valutazione finora
- 435 Note 2Documento37 pagine435 Note 2PreciousNessuna valutazione finora
- Thermodynamics CompleteDocumento43 pagineThermodynamics Completesutarohit2006Nessuna valutazione finora
- Simple Applications of Macroscopic ThermodynamicsDocumento33 pagineSimple Applications of Macroscopic ThermodynamicsreianreyNessuna valutazione finora
- Chengbo WangDocumento5 pagineChengbo WangWalid MohamedNessuna valutazione finora
- 5-Summary For Lectures 1,2,3,4Documento17 pagine5-Summary For Lectures 1,2,3,4losblancos1Nessuna valutazione finora
- Exam Thermo Part1!11!12 2020 FinalDocumento11 pagineExam Thermo Part1!11!12 2020 FinalMaarten ElingNessuna valutazione finora
- Thermo2 PDFDocumento12 pagineThermo2 PDFBreezyNessuna valutazione finora
- CH 11Documento14 pagineCH 11hirenpatel_universalNessuna valutazione finora
- Joule Thomson EffectDocumento11 pagineJoule Thomson EffectEdmond YuenNessuna valutazione finora
- ATAUKSSDocumento10 pagineATAUKSSbullo hindebuNessuna valutazione finora
- CFD Lecture 1Documento8 pagineCFD Lecture 1ethiopian art2020Nessuna valutazione finora
- l3 Thermo 23Documento83 paginel3 Thermo 23leomessi73870Nessuna valutazione finora
- Derivation of Navier Stokes Momentum EquationsDocumento2 pagineDerivation of Navier Stokes Momentum EquationsFbgames StefNessuna valutazione finora
- 2023 CIR310 Sem Test 1 With AnswersDocumento7 pagine2023 CIR310 Sem Test 1 With Answersu21589969Nessuna valutazione finora
- MAT397 SP 11 Practice Exam 2 SolutionsDocumento7 pagineMAT397 SP 11 Practice Exam 2 SolutionsRuben VelasquezNessuna valutazione finora
- NST Mmii Chapter2Documento27 pagineNST Mmii Chapter2Diego Canales AguileraNessuna valutazione finora
- School of Physics and Astronomy: File Topic09 PDFDocumento4 pagineSchool of Physics and Astronomy: File Topic09 PDFzjnsrbtNessuna valutazione finora
- Điều Kiện Phương Trình Virial Hệ Số Thứ 2Documento17 pagineĐiều Kiện Phương Trình Virial Hệ Số Thứ 2Marcus NguyễnNessuna valutazione finora
- Review of Thermodynamics and Intro To Statistical Mechanics: Prof. Mark W. Tibbitt - ETH Z Urich - 21 Februar 2019Documento9 pagineReview of Thermodynamics and Intro To Statistical Mechanics: Prof. Mark W. Tibbitt - ETH Z Urich - 21 Februar 2019Luis Fernando RodriguezNessuna valutazione finora
- DiffgeoDocumento20 pagineDiffgeoMANIRAGUHA Jean PaulNessuna valutazione finora
- Esci342 Lesson07 Continuity EquationDocumento8 pagineEsci342 Lesson07 Continuity EquationMohamed Abd El-MoniemNessuna valutazione finora
- Partial Diffrential EquationDocumento29 paginePartial Diffrential Equationعلي عثمانNessuna valutazione finora
- 2021 Geometry Study Guide 3Documento15 pagine2021 Geometry Study Guide 3JacmNessuna valutazione finora
- Uni of Frankfurt - Thermodynamic PotentialsDocumento15 pagineUni of Frankfurt - Thermodynamic PotentialstaboogaNessuna valutazione finora
- Hemodynamics Notes: 1 Navier Stokes DerivationDocumento3 pagineHemodynamics Notes: 1 Navier Stokes DerivationAndrew ScharfNessuna valutazione finora
- Green's Function Estimates for Lattice Schrödinger Operators and Applications. (AM-158)Da EverandGreen's Function Estimates for Lattice Schrödinger Operators and Applications. (AM-158)Nessuna valutazione finora
- Fundamentals of NetworksDocumento80 pagineFundamentals of NetworksSamriddha Das GuptaNessuna valutazione finora
- Course IntroductionDocumento16 pagineCourse IntroductionSamriddha Das GuptaNessuna valutazione finora
- Laboratory Manual MS Sem VDocumento32 pagineLaboratory Manual MS Sem VSamriddha Das GuptaNessuna valutazione finora
- Institute of Technology: Course PolicyDocumento8 pagineInstitute of Technology: Course PolicySamriddha Das GuptaNessuna valutazione finora
- Introduction To MATLABDocumento47 pagineIntroduction To MATLABSamriddha Das GuptaNessuna valutazione finora
- Introduction To Process Simulators and Process SimulationDocumento12 pagineIntroduction To Process Simulators and Process SimulationSamriddha Das GuptaNessuna valutazione finora
- Time Value of MoneyDocumento10 pagineTime Value of MoneySamriddha Das GuptaNessuna valutazione finora
- CPI Virtual Lab: Presented By: S Amriddha Das Gupta (18BCH055)Documento36 pagineCPI Virtual Lab: Presented By: S Amriddha Das Gupta (18BCH055)Samriddha Das GuptaNessuna valutazione finora
- Environment IntroductionDocumento10 pagineEnvironment IntroductionSamriddha Das GuptaNessuna valutazione finora
- IPC Term Paper: Presented By: Samriddha Das Gupta (18BCH055)Documento28 pagineIPC Term Paper: Presented By: Samriddha Das Gupta (18BCH055)Samriddha Das GuptaNessuna valutazione finora
- Lab Manuals 2CH403 IPC Jan 2020 PDFDocumento47 pagineLab Manuals 2CH403 IPC Jan 2020 PDFSamriddha Das GuptaNessuna valutazione finora
- CPI Virtual Lab: Present Ed By: Samriddha Das Gupta (18BCH055)Documento36 pagineCPI Virtual Lab: Present Ed By: Samriddha Das Gupta (18BCH055)Samriddha Das GuptaNessuna valutazione finora
- PC 1 PDFDocumento27 paginePC 1 PDFSamriddha Das GuptaNessuna valutazione finora
- Process Heat Transfer Hof MasterDocumento156 pagineProcess Heat Transfer Hof MasterbadelitamariusNessuna valutazione finora
- Casing Design 3Documento32 pagineCasing Design 3Anes dzNessuna valutazione finora
- Chemical EquilibriumDocumento49 pagineChemical EquilibriumPhoemela SangumayNessuna valutazione finora
- 978 981 15 5753 8 - 50Documento15 pagine978 981 15 5753 8 - 50Chukwumaobi OluahNessuna valutazione finora
- Etabs 2016 16.2Documento6 pagineEtabs 2016 16.2kervinarmasNessuna valutazione finora
- CHAPTER 6 Particle TheoryDocumento7 pagineCHAPTER 6 Particle TheoryEunice Xiiao WennNessuna valutazione finora
- Chapter 2Documento10 pagineChapter 2abdul azizNessuna valutazione finora
- Chapter 8-Filtration (56 P)Documento55 pagineChapter 8-Filtration (56 P)shardulkaviNessuna valutazione finora
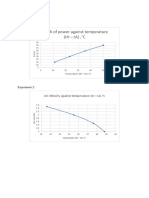
- Graph of Power Against Temperature (TH - Ta) ,°C: Experiment 1Documento3 pagineGraph of Power Against Temperature (TH - Ta) ,°C: Experiment 1Muhd UbaiNessuna valutazione finora
- CLT2Documento13 pagineCLT2Yagnik KalariyaNessuna valutazione finora
- Johnson Cook StrengthDocumento2 pagineJohnson Cook StrengthClaudio Martín GimenezNessuna valutazione finora
- Experimental and Numerical Studies On Geotextile Reinforced Subgrade SoilDocumento14 pagineExperimental and Numerical Studies On Geotextile Reinforced Subgrade SoilweeerieNessuna valutazione finora
- Gas Diffusion UnitDocumento20 pagineGas Diffusion Unitsolehah misniNessuna valutazione finora
- Composite and Non-Composite Behaviors of Foam-Insulated Concrete Sandwich PanelsDocumento9 pagineComposite and Non-Composite Behaviors of Foam-Insulated Concrete Sandwich Panelstimtoihochoi1Nessuna valutazione finora
- Bellary2015 - RugosidadDocumento11 pagineBellary2015 - RugosidadAbraham SilesNessuna valutazione finora
- 026 PDFDocumento11 pagine026 PDFMEHDINessuna valutazione finora
- Steam Jet RefrigerationDocumento14 pagineSteam Jet RefrigerationSAATVIK JAINNessuna valutazione finora
- Part A - Part A Question No.1 Bookmark: (Chosen Option)Documento27 paginePart A - Part A Question No.1 Bookmark: (Chosen Option)selvakumarNessuna valutazione finora
- Temperature MeasurementDocumento57 pagineTemperature MeasurementATUL ASWARNessuna valutazione finora