Potrebbero piacerti anche
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (120)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Full Body Workout B PDFDocumento17 pagineFull Body Workout B PDFDiogo Proença74% (23)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- ChemistryDocumento417 pagineChemistryKetsya Lenak100% (2)
- Secondary 2 Science NotesDocumento43 pagineSecondary 2 Science Noteszach0% (1)
- Shanghai Singapore International School G11 Chemistry Test - Bonding SL Mark Out of .. Name Target Grade . Best Grade 1Documento10 pagineShanghai Singapore International School G11 Chemistry Test - Bonding SL Mark Out of .. Name Target Grade . Best Grade 1oscarbecNessuna valutazione finora
- Test Bank For Organic Chemistry 7th Edition by L G Wade JR Test BankDocumento31 pagineTest Bank For Organic Chemistry 7th Edition by L G Wade JR Test BankHarold Welborn100% (36)
- Answer KEY Set ADocumento1 paginaAnswer KEY Set Aratnesh200Nessuna valutazione finora
- Answer KEY Set ADocumento1 paginaAnswer KEY Set Aratnesh200Nessuna valutazione finora
- Normal ShocDocumento12 pagineNormal Shocratnesh200Nessuna valutazione finora
- UnfairmeansDocumento1 paginaUnfairmeansratnesh200Nessuna valutazione finora
- Tut 13Documento1 paginaTut 13ratnesh200Nessuna valutazione finora
- Answer KEY Set ADocumento1 paginaAnswer KEY Set Aratnesh200Nessuna valutazione finora
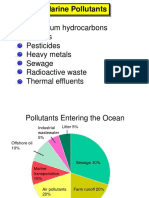
- Lecture 10 - BITS F225 - Marine Pollution PDFDocumento24 pagineLecture 10 - BITS F225 - Marine Pollution PDFratnesh200Nessuna valutazione finora
- Tut 12 PDFDocumento1 paginaTut 12 PDFratnesh200Nessuna valutazione finora
- Tut 12 PDFDocumento1 paginaTut 12 PDFratnesh200Nessuna valutazione finora
- Lecture 9 - BITS F225 - Air Pollution PDFDocumento22 pagineLecture 9 - BITS F225 - Air Pollution PDFratnesh200Nessuna valutazione finora
- Tut 11Documento1 paginaTut 11ratnesh200Nessuna valutazione finora
- Lecture4 Characteristics of Covalent BondDocumento33 pagineLecture4 Characteristics of Covalent BondMikhailah SigangNessuna valutazione finora
- Electron Counting Methods 1) Neutral Ligand / Covalent MethodDocumento8 pagineElectron Counting Methods 1) Neutral Ligand / Covalent MethodRakshitTiwariNessuna valutazione finora
- The Kinetic Molecular Model and Intermolecular Forces of Attraction in MatterDocumento104 pagineThe Kinetic Molecular Model and Intermolecular Forces of Attraction in MatterMiguel TatlonghariNessuna valutazione finora
- 11 Chemistry Notes Ch09 HydrogenDocumento4 pagine11 Chemistry Notes Ch09 HydrogenTanisha MoriNessuna valutazione finora
- Practice Chap 1 and 2 - AnswerDocumento11 paginePractice Chap 1 and 2 - AnswerNur Afiqah Mohd ZakiNessuna valutazione finora
- Free Energy-Fact or Fiction?: by Maurice CotterellDocumento4 pagineFree Energy-Fact or Fiction?: by Maurice Cotterellfsilassie8012Nessuna valutazione finora
- Carbon and Its Compounds Lesson Notes TS SSCDocumento35 pagineCarbon and Its Compounds Lesson Notes TS SSCSAI PRANEETH REDDY DHADINessuna valutazione finora
- 8.2 Chemical Earth NotesDocumento14 pagine8.2 Chemical Earth NotesCatherine Lai100% (6)
- Crystal Chemistry NotesDocumento37 pagineCrystal Chemistry NotesdivyaNessuna valutazione finora
- Chemistry Revision Work Grade 10 To 12Documento127 pagineChemistry Revision Work Grade 10 To 12samora malungisaNessuna valutazione finora
- HSSRPTR - +1 Chemistry Focus Area NotesDocumento58 pagineHSSRPTR - +1 Chemistry Focus Area NotesAbduk100% (2)
- Carbon and Its Compounds NotesDocumento27 pagineCarbon and Its Compounds NotesBALAJI VARA PRASAD100% (1)
- Quiz G9 ScienceDocumento1 paginaQuiz G9 ScienceJessica CorpuzNessuna valutazione finora
- Course Planner Ajay Phase ResonanceDocumento4 pagineCourse Planner Ajay Phase ResonanceMAHA BHARATNessuna valutazione finora
- GRP 17 - P Block ChemhackDocumento7 pagineGRP 17 - P Block ChemhackYuvarajNessuna valutazione finora
- Feb 2 ReflectionDocumento4 pagineFeb 2 Reflection07232017Nessuna valutazione finora
- 10th Science Practice TestDocumento13 pagine10th Science Practice Testavinash960Nessuna valutazione finora
- CHM 115 Lecture NotesDocumento4 pagineCHM 115 Lecture NotesHao ZhangNessuna valutazione finora
- GGPS Syllabus 11Documento42 pagineGGPS Syllabus 11tajbgpNessuna valutazione finora
- Average Values From Many Compounds Used in BondDocumento67 pagineAverage Values From Many Compounds Used in BondG M Ali KawsarNessuna valutazione finora
- Essentials of Materials Science and Engineering Si Edition 3rd Edition Askeland Solutions ManualDocumento11 pagineEssentials of Materials Science and Engineering Si Edition 3rd Edition Askeland Solutions Manualjeffreyhayesagoisypdfm100% (13)
- Ionic and Covalent BondsDocumento5 pagineIonic and Covalent BondsFern HofileñaNessuna valutazione finora
- Gravitational and Gravity Constant, and Their Physical Interpretation .Documento73 pagineGravitational and Gravity Constant, and Their Physical Interpretation .georgallidesmarcosNessuna valutazione finora
- S-Blockelements PDFDocumento88 pagineS-Blockelements PDFchingy100% (2)
- Chemistry Module 2 - Part 2Documento13 pagineChemistry Module 2 - Part 2Francis RecocoNessuna valutazione finora
- Crystal and Dislocations: StructureDocumento148 pagineCrystal and Dislocations: StructureTulasidas MalajiNessuna valutazione finora