Potrebbero piacerti anche
- Sistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Da EverandSistema nervioso y osteopatía: Nervios periféricos, meninges craneales y espinales, y sistema nervioso vegetativo (Color)Valutazione: 5 su 5 stelle5/5 (9)
- Electromiografia en Rehabilitacion2Documento50 pagineElectromiografia en Rehabilitacion2Andres Florian Florian100% (1)
- ELA NEUROFISIOLOGIADocumento25 pagineELA NEUROFISIOLOGIAfaquipe007Nessuna valutazione finora
- Neuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaDa EverandNeuroanatomía: Fundamentos de neuroanatomía estructural, funcional y clínicaValutazione: 4 su 5 stelle4/5 (16)
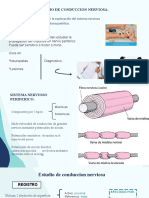
- Estudios de Conduccion NerviosaDocumento15 pagineEstudios de Conduccion NerviosaJose la CruzNessuna valutazione finora
- Tendón: Valoración y tratamiento en fisioterapiaDa EverandTendón: Valoración y tratamiento en fisioterapiaValutazione: 5 su 5 stelle5/5 (4)
- Sem - Debilidad MuscularDocumento21 pagineSem - Debilidad MuscularArnovioNessuna valutazione finora
- Mioclonías: Características Clínicas y Neurofisiológicas, Etiología y TratamientoDocumento6 pagineMioclonías: Características Clínicas y Neurofisiológicas, Etiología y TratamientoJohn AriasNessuna valutazione finora
- Cap 34Documento30 pagineCap 34Annette MolinaNessuna valutazione finora
- ElaDocumento2 pagineElaMelany MedinaNessuna valutazione finora
- EMG en EsquemasDocumento4 pagineEMG en EsquemasFlorencia MercadoNessuna valutazione finora
- MotoneuronaDocumento16 pagineMotoneuronaSilvy FuentesNessuna valutazione finora
- Evaluación de pacientes con enfermedades neuromuscularesDocumento6 pagineEvaluación de pacientes con enfermedades neuromuscularesNury Pabón CastilloNessuna valutazione finora
- Amiotrofias Espinales ProgresivasDocumento18 pagineAmiotrofias Espinales Progresivasapi-3814371Nessuna valutazione finora
- Esclerosis Lateral AmiotroficaDocumento4 pagineEsclerosis Lateral Amiotroficaalphonse floraNessuna valutazione finora
- Neurona Motora Inferior (Nmi)Documento13 pagineNeurona Motora Inferior (Nmi)Johanna GalvisNessuna valutazione finora
- Enfermedades de las motoneuronas: actualización sobre ELA y AMEDocumento11 pagineEnfermedades de las motoneuronas: actualización sobre ELA y AMEAngie marcela Ortiz carrascalNessuna valutazione finora
- Enfermedades de La MotoneuronaDocumento11 pagineEnfermedades de La MotoneuronaLAURA PEREIRA CERONNessuna valutazione finora
- Neuropatia Diabetica. Della Nina, DafneDocumento11 pagineNeuropatia Diabetica. Della Nina, DafneAlma DonnaNessuna valutazione finora
- Esclerosis Lateral Amiotrófica: Enfermedad Neurológica Progresiva e Invariablemente FatalDocumento16 pagineEsclerosis Lateral Amiotrófica: Enfermedad Neurológica Progresiva e Invariablemente FatalVerónica AvilaNessuna valutazione finora
- Tema 51 Función Motora FINALDocumento10 pagineTema 51 Función Motora FINALLuYsLuNessuna valutazione finora
- 10.1016@B978 0 444 64142 7.00054 0Documento10 pagine10.1016@B978 0 444 64142 7.00054 0Ricardo Peraza RiveroNessuna valutazione finora
- Debilidad MuscularDocumento17 pagineDebilidad MuscularArnovioNessuna valutazione finora
- Mononeuropatia MúltipleDocumento27 pagineMononeuropatia MúltiplesolecitodelmarNessuna valutazione finora
- Esclerosis Lateral AmiotróficaDocumento3 pagineEsclerosis Lateral AmiotróficaJUAN CHAVEZNessuna valutazione finora
- Electromiografia: Prof. Angela SantorsolaDocumento43 pagineElectromiografia: Prof. Angela SantorsolaManuel MoraNessuna valutazione finora
- Enfermedad de la motoneurona: síndromes y clasificaciónDocumento9 pagineEnfermedad de la motoneurona: síndromes y clasificaciónLizethNessuna valutazione finora
- Ela-Grupo ViiiDocumento17 pagineEla-Grupo ViiiAlonso Sánchez DuránNessuna valutazione finora
- ELA Esclerosis Lateral AmiotróficaDocumento20 pagineELA Esclerosis Lateral AmiotróficaFrancisco ChalenNessuna valutazione finora
- Trabajo EscritoDocumento9 pagineTrabajo EscritoIxba GGNessuna valutazione finora
- Sindrome de Motoneurona InferiorDocumento29 pagineSindrome de Motoneurona InferiorJenny AmasthaNessuna valutazione finora
- Trauma Medular (Impreso)Documento24 pagineTrauma Medular (Impreso)Paulina Constanza López PinedaNessuna valutazione finora
- Enfermedad de ParkinsonDocumento10 pagineEnfermedad de ParkinsonLiseth Paola Sierra Anachury100% (1)
- Enfermedades de La Neurona MotoraDocumento39 pagineEnfermedades de La Neurona MotoraAbraham GonzálezNessuna valutazione finora
- Enfermedades neuronas motoras superiores e inferioresDocumento4 pagineEnfermedades neuronas motoras superiores e inferioresLuisa ReyesNessuna valutazione finora
- Neuromiotonía y mioquimia: definición y manifestaciones clínicasDocumento8 pagineNeuromiotonía y mioquimia: definición y manifestaciones clínicaseri suyoNessuna valutazione finora
- 23-10 SINDROME HIPOTONICO-imprimirDocumento36 pagine23-10 SINDROME HIPOTONICO-imprimirapi-3740897100% (6)
- Cómo y Cuándo - Estudios ElectrodiagnósticoDocumento5 pagineCómo y Cuándo - Estudios ElectrodiagnósticoSantiago Andres Gomez MorenoNessuna valutazione finora
- Electro en Cefalo GramaDocumento67 pagineElectro en Cefalo GramaCastillo Alejandre CarlosNessuna valutazione finora
- Debilidad Aguda SimetricaDocumento12 pagineDebilidad Aguda SimetricaOmar Cristian Sanchez100% (1)
- ELECTROMIOGRAFÍADocumento15 pagineELECTROMIOGRAFÍAalbertwillyNessuna valutazione finora
- Neuropatias 2019.Ppt - DegrabadaDocumento108 pagineNeuropatias 2019.Ppt - DegrabadaLuis Yehsuah Moran AmayaNessuna valutazione finora
- Exposicion y Algoritmo Segunda MotoneuronaDocumento29 pagineExposicion y Algoritmo Segunda MotoneuronaErick AñarumbaNessuna valutazione finora
- Actividad 1 Unidad 3 NeurocienciasDocumento3 pagineActividad 1 Unidad 3 NeurocienciasAngel Cortez MendivilNessuna valutazione finora
- Ela 2Documento4 pagineEla 2Yessenia Ramirez AlvaradoNessuna valutazione finora
- Cap 10Documento4 pagineCap 10Adrian ChaviraNessuna valutazione finora
- Exposición de NeurologiaDocumento17 pagineExposición de NeurologiaSHEYLA PRISSILA TRUJILLO VARGASNessuna valutazione finora
- Motoneurona CompletoDocumento12 pagineMotoneurona Completomayaecheverria9Nessuna valutazione finora
- 8 Electrodiagnostico Electromiografia 100626112455 Phpapp02 PDFDocumento3 pagine8 Electrodiagnostico Electromiografia 100626112455 Phpapp02 PDFFabian Esteban SubiabreNessuna valutazione finora
- Anotaciones PRÁCTICA 03Documento5 pagineAnotaciones PRÁCTICA 03KIMBERLY BRISSETH CIEZA VARGASNessuna valutazione finora
- Musculo NeuroDocumento57 pagineMusculo Neuroelivi78Nessuna valutazione finora
- Sme Piramidal - Alteraciones de La SensibilidadDocumento3 pagineSme Piramidal - Alteraciones de La SensibilidadFiamma SassiNessuna valutazione finora
- DiagnósticoDocumento23 pagineDiagnósticoTatiana FuenmayorNessuna valutazione finora
- Sindrome de Neurona MotoraDocumento2 pagineSindrome de Neurona MotoraYahaira Borquez RiosNessuna valutazione finora
- SX Neurona Motora InferiorDocumento27 pagineSX Neurona Motora InferiorBaushell B. KingNessuna valutazione finora
- Esclerosis Lateral AmiotroficaDocumento10 pagineEsclerosis Lateral AmiotroficaMilton MoscosoNessuna valutazione finora
- Enf. NeuromuscularesDocumento7 pagineEnf. NeuromuscularesPamela Giust100% (1)
- Expo NeuronaDocumento19 pagineExpo NeuronaMendoza Ramírez RossNessuna valutazione finora
- Expo Final - Teoría G2Documento18 pagineExpo Final - Teoría G2Silvia Eche echeNessuna valutazione finora
- ELA y Estudios NeurofisiológicosDocumento10 pagineELA y Estudios Neurofisiológicosanleegonzalezg80 ggNessuna valutazione finora
- 528 Paralisis FacialDocumento8 pagine528 Paralisis FacialVanesa TiRiaNessuna valutazione finora
- Transferencia Nerviosa o NeurotizaciónDocumento1 paginaTransferencia Nerviosa o Neurotizaciónanleegonzalezg80 ggNessuna valutazione finora
- Salida Toracica PDFDocumento12 pagineSalida Toracica PDFanleegonzalezg80 ggNessuna valutazione finora
- Rehabilitación de Pacientes HemipléjicosDocumento6 pagineRehabilitación de Pacientes HemipléjicosMac Perez LopezNessuna valutazione finora
- Síndrome Tunel TarsianoDocumento2 pagineSíndrome Tunel Tarsianoanleegonzalezg80 ggNessuna valutazione finora
- PolineuropatiasDocumento18 paginePolineuropatiasanleegonzalezg80 ggNessuna valutazione finora
- Derecho Penal Primera FaseDocumento81 pagineDerecho Penal Primera FaseRodrio NeiraNessuna valutazione finora
- Plan integral seguridad PopayánDocumento6 paginePlan integral seguridad PopayánJhoanaAndreaMuñozNessuna valutazione finora
- Imagenologia en UrologiaDocumento78 pagineImagenologia en UrologiaPatricia victoriaNessuna valutazione finora
- Anastomosis IntestinalDocumento55 pagineAnastomosis Intestinalgeorge66500Nessuna valutazione finora
- Proyecto Final EnviarDocumento85 pagineProyecto Final Enviarclaudia camposNessuna valutazione finora
- ÉticaDocumento7 pagineÉticafanny leonor prado martinezNessuna valutazione finora
- Proyecto Como Influyen Las Tic en Las Plantas MedicinalesDocumento9 pagineProyecto Como Influyen Las Tic en Las Plantas MedicinalesAnonymous N1437t7GNessuna valutazione finora
- Escala de Actitud Hacia La HomosexualidadDocumento3 pagineEscala de Actitud Hacia La HomosexualidadJuan CarlosNessuna valutazione finora
- R13 VIRUS Y VIROIDES Intro A FitoDocumento4 pagineR13 VIRUS Y VIROIDES Intro A FitoWiliam Ixpatá IxtecocNessuna valutazione finora
- ANALISISDocumento8 pagineANALISISKaren QuiguagoNessuna valutazione finora
- Exp-Pln-004 Procedimiento Limpieza de Pozas de SedimentacionDocumento5 pagineExp-Pln-004 Procedimiento Limpieza de Pozas de SedimentacionJulio RamiroNessuna valutazione finora
- VITA 1963 1963SP VITA MFT Aufsteller VA ES V02 Screen EsDocumento32 pagineVITA 1963 1963SP VITA MFT Aufsteller VA ES V02 Screen EsAlexander SotomayorNessuna valutazione finora
- Togavirus y FlavivirusDocumento4 pagineTogavirus y FlavivirusyunniyomiNessuna valutazione finora
- Listado Prestadores Definitivo-19Documento10 pagineListado Prestadores Definitivo-19Marcos PereaNessuna valutazione finora
- Proyecto Dada Libre de HumoDocumento14 pagineProyecto Dada Libre de HumoCesar RivasNessuna valutazione finora
- IRA Tipo I: Caso Clínico de Paciente con Insuficiencia Respiratoria AgudaDocumento43 pagineIRA Tipo I: Caso Clínico de Paciente con Insuficiencia Respiratoria AgudaLinder Tapia ChumbeNessuna valutazione finora
- ZleepDocumento24 pagineZleepLusdoralvis Jose Malave PerezNessuna valutazione finora
- Expo Psi Cote Rap 811Documento51 pagineExpo Psi Cote Rap 811Ger BrennerNessuna valutazione finora
- El Reforma 09-12-20Documento50 pagineEl Reforma 09-12-20enock-readersNessuna valutazione finora
- Procedimiento seguro winche arrastreDocumento5 pagineProcedimiento seguro winche arrastreCesarRamirezCNessuna valutazione finora
- LA SEXUALIDAD A LO LARGO DE LA HISTORIA MañanaDocumento8 pagineLA SEXUALIDAD A LO LARGO DE LA HISTORIA MañanaNataly AbyNessuna valutazione finora
- Factibilidad Moringa PDFDocumento178 pagineFactibilidad Moringa PDFJose Antonio Guerrero FLoresNessuna valutazione finora
- Línea esmalte CERELUXE HDSDocumento5 pagineLínea esmalte CERELUXE HDSRodrigo Alberto Bermúdez Pérez33% (3)
- Año Del Fortalecimiento de La Soberania NacionalDocumento16 pagineAño Del Fortalecimiento de La Soberania NacionalYamil Dominguez LopezNessuna valutazione finora
- Plan de Emergencias RadiológicasDocumento25 paginePlan de Emergencias Radiológicasjgaravi1100% (1)
- Actividad 2 Gasolinera La MorteraDocumento5 pagineActividad 2 Gasolinera La MorteraMonica Andrea Gomez FlorNessuna valutazione finora
- 16-Texto Del Artículo-79-1-10-20200826Documento10 pagine16-Texto Del Artículo-79-1-10-20200826Luis Antonio Sánchez SánchezNessuna valutazione finora
- Antiinflamatorios esteroides: dexametasonaDocumento40 pagineAntiinflamatorios esteroides: dexametasonaOinotna Zelaznog Zetineb87% (47)
- Guia Practica - Unidad 02Documento11 pagineGuia Practica - Unidad 02Adriana CarrascoNessuna valutazione finora
- Neumoconiosis: causas, síntomas y prevención de esta enfermedad laboralDocumento9 pagineNeumoconiosis: causas, síntomas y prevención de esta enfermedad laboralYuly Dayana GARCIA PERILLANessuna valutazione finora
- El Monje Que Vendio Su Ferrari: Una Fábula EspiritualDa EverandEl Monje Que Vendio Su Ferrari: Una Fábula EspiritualValutazione: 4.5 su 5 stelle4.5/5 (1695)
- Las seis etapas definitivas para superar tu divorcioDa EverandLas seis etapas definitivas para superar tu divorcioValutazione: 4.5 su 5 stelle4.5/5 (10)
- Tu cerebro emocional: Saca partido de lo que sientes y transforma tu vidaDa EverandTu cerebro emocional: Saca partido de lo que sientes y transforma tu vidaValutazione: 5 su 5 stelle5/5 (2)
- Resetea tu mente. Descubre de lo que eres capazDa EverandResetea tu mente. Descubre de lo que eres capazValutazione: 5 su 5 stelle5/5 (196)
- Batidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoDa EverandBatidos Verdes Depurativos y Antioxidantes: Aumenta tu Vitalidad con Smoothie Detox Durante 10 Días Para Adelgazar y Bajar de Peso: Aumenta tu vitalidad con smoothie detox durante 10 días para adelgazar y bajar de pesoValutazione: 5 su 5 stelle5/5 (2)
- ¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadDa Everand¡Tómate un respiro! Mindfulness: El arte de mantener la calma en medio de la tempestadValutazione: 5 su 5 stelle5/5 (198)
- Cómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaDa EverandCómo hacer que te pasen cosas buenas: Entiende tu cerebro, gestiona tus emociones, mejora tu vidaValutazione: 5 su 5 stelle5/5 (1866)
- La revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaDa EverandLa revolución de la glucosa: Equilibra tus niveles de glucosa y cambiarás tu salud y tu vidaValutazione: 5 su 5 stelle5/5 (200)
- Los 12 chakras: Desbloquea tus dones espiritualesDa EverandLos 12 chakras: Desbloquea tus dones espiritualesValutazione: 4 su 5 stelle4/5 (14)
- Disciplina con amor para adolescentes (Discipline With Love for Adolescents): Guía para llevarte bien con tu adolescente (A Guide for Getting Along Well With Your Adolescent)Da EverandDisciplina con amor para adolescentes (Discipline With Love for Adolescents): Guía para llevarte bien con tu adolescente (A Guide for Getting Along Well With Your Adolescent)Valutazione: 5 su 5 stelle5/5 (10)
- El poder del optimismo: Herramientas para vivir de forma más positivaDa EverandEl poder del optimismo: Herramientas para vivir de forma más positivaValutazione: 4.5 su 5 stelle4.5/5 (15)
- Terapia cognitivo-conductual (TCC) y terapia dialéctico-conductual (TDC): Cómo la TCC, la TDC y la ACT pueden ayudarle a superar la ansiedad, la depresión, y los TOCSDa EverandTerapia cognitivo-conductual (TCC) y terapia dialéctico-conductual (TDC): Cómo la TCC, la TDC y la ACT pueden ayudarle a superar la ansiedad, la depresión, y los TOCSValutazione: 5 su 5 stelle5/5 (1)
- Viaje a través del libro de ejercicios de un curso de milagros. Volumen 1Da EverandViaje a través del libro de ejercicios de un curso de milagros. Volumen 1Valutazione: 4 su 5 stelle4/5 (8)
- LOS ARCANOS DE NACIMIENTO: EL TAROT DEL ALMA: ¿CÓMO CALCULAR TU ARCANO PERSONAL O DE ALMA?Da EverandLOS ARCANOS DE NACIMIENTO: EL TAROT DEL ALMA: ¿CÓMO CALCULAR TU ARCANO PERSONAL O DE ALMA?Valutazione: 5 su 5 stelle5/5 (8)
- Influencia. La psicología de la persuasiónDa EverandInfluencia. La psicología de la persuasiónValutazione: 4.5 su 5 stelle4.5/5 (14)
- La Tabla Esmeralda: Incluye varias versiones y explicacionesDa EverandLa Tabla Esmeralda: Incluye varias versiones y explicacionesValutazione: 4.5 su 5 stelle4.5/5 (7)
- ¿Por qué mis padres no me aman?: Empezando a sanarDa Everand¿Por qué mis padres no me aman?: Empezando a sanarValutazione: 4.5 su 5 stelle4.5/5 (33)
- Resumen de El Sutil Arte de que te Importe un Carajo, de Mark MansonDa EverandResumen de El Sutil Arte de que te Importe un Carajo, de Mark MansonValutazione: 4.5 su 5 stelle4.5/5 (15)
- ¡Basta ya de ser un Tipo Lindo! (No More Mr. Nice Guy): Un Plan Probado para Que Obtengas Lo Que Quieras en La Vida El Sexo y El Amor (A Proven Plan for Getting What You Want in Love, Sex and Life)Da Everand¡Basta ya de ser un Tipo Lindo! (No More Mr. Nice Guy): Un Plan Probado para Que Obtengas Lo Que Quieras en La Vida El Sexo y El Amor (A Proven Plan for Getting What You Want in Love, Sex and Life)Valutazione: 5 su 5 stelle5/5 (47)
- Despierta tu Energía Femenina: Secretos de Energía de la Diosa y Cómo Acceder a Tu Poder DivinoDa EverandDespierta tu Energía Femenina: Secretos de Energía de la Diosa y Cómo Acceder a Tu Poder DivinoValutazione: 4 su 5 stelle4/5 (5)
- Psiconeuroinmunología para la práctica clínicaDa EverandPsiconeuroinmunología para la práctica clínicaValutazione: 5 su 5 stelle5/5 (4)