Potrebbero piacerti anche
- Rare Genital Disseminated Superficial Porokeratosis in Immunocompetent ManDocumento3 pagineRare Genital Disseminated Superficial Porokeratosis in Immunocompetent ManMostafaAhmedNessuna valutazione finora
- Collodion Baby: Linical CaseDocumento7 pagineCollodion Baby: Linical CaseDyah Farah DetaNessuna valutazione finora
- Canine Ichthyosis and Related Disorders of CornificationDocumento9 pagineCanine Ichthyosis and Related Disorders of CornificationPaolaNessuna valutazione finora
- Keratosis Pilaris and Darier DiseaseDocumento9 pagineKeratosis Pilaris and Darier DiseaseMaulana RamziNessuna valutazione finora
- Nelson Adesokan1992Documento6 pagineNelson Adesokan1992Yash PatilNessuna valutazione finora
- Herediter and Acquired Ichthyosis VulgarisDocumento4 pagineHerediter and Acquired Ichthyosis VulgarisprajnamitaNessuna valutazione finora
- CADAE1405CDocumento12 pagineCADAE1405CMuhammad RiduanNessuna valutazione finora
- Ichthyosis: A Road Model For Skin Research: Review ArticleDocumento10 pagineIchthyosis: A Road Model For Skin Research: Review ArticlebaihaqiNessuna valutazione finora
- Cornoid Lamellation Associated With Lichen SclerosusDocumento2 pagineCornoid Lamellation Associated With Lichen SclerosusJay GHelaNessuna valutazione finora
- Dermatologia: Anais Brasileiros deDocumento3 pagineDermatologia: Anais Brasileiros deSango AyraNessuna valutazione finora
- Squamous Epithelial Changes of The Larynx: Diagnosis and TherapyDocumento8 pagineSquamous Epithelial Changes of The Larynx: Diagnosis and TherapyIsrael BlancoNessuna valutazione finora
- X-Linked Incontinentia Pigmenti or Bloch-Sulzberger Syndrome: A Case ReportDocumento4 pagineX-Linked Incontinentia Pigmenti or Bloch-Sulzberger Syndrome: A Case ReportOman ArifNessuna valutazione finora
- Genetic Molecular Basis of Cutis Laxa and Surgical ManagementDocumento10 pagineGenetic Molecular Basis of Cutis Laxa and Surgical ManagementIOSRjournalNessuna valutazione finora
- Ajd v1 Id1016Documento3 pagineAjd v1 Id1016Ari KurniawanNessuna valutazione finora
- Berk 2012Documento17 pagineBerk 2012abdoh hassanNessuna valutazione finora
- UntitledDocumento3 pagineUntitledShintaNessuna valutazione finora
- Folliculitis Decalvans: The Use of Dermatoscopy As An Auxiliary Tool in Clinical DiagnosisDocumento3 pagineFolliculitis Decalvans: The Use of Dermatoscopy As An Auxiliary Tool in Clinical DiagnosisKatherine PatiñoNessuna valutazione finora
- Cryptorchidism and Its Long-Term ComplicationsDocumento6 pagineCryptorchidism and Its Long-Term ComplicationsYacine Tarik AizelNessuna valutazione finora
- Keloids: Clinica L Fea Tures A ND M A Na Gem Ent.: Review ArticleDocumento8 pagineKeloids: Clinica L Fea Tures A ND M A Na Gem Ent.: Review ArticleAndri Feisal NasutionNessuna valutazione finora
- Ichthyosis-Characteristic: Appearance-Mental Retardation Syndrome With Distinct Histological Skin AbnormalitiesDocumento4 pagineIchthyosis-Characteristic: Appearance-Mental Retardation Syndrome With Distinct Histological Skin AbnormalitiesJoNessuna valutazione finora
- Spindle Cell Lipoma: Franz Enzinger, A. HarveyDocumento8 pagineSpindle Cell Lipoma: Franz Enzinger, A. HarveyAdjhy Aji AchmadNessuna valutazione finora
- Collodion baby: Practical management and outcomesDocumento13 pagineCollodion baby: Practical management and outcomesHend AbdallaNessuna valutazione finora
- Treachercollinssyndrome: Albaraa Aljerian,, Mirko S. GilardinoDocumento9 pagineTreachercollinssyndrome: Albaraa Aljerian,, Mirko S. GilardinoSai KrupaNessuna valutazione finora
- The Ichthyosis Encyclopedia: Tests, Causes and TreatmentsDa EverandThe Ichthyosis Encyclopedia: Tests, Causes and TreatmentsNessuna valutazione finora
- International Nosology Heritable Disorders of Connective Tissue, Berlin, 1986Documento14 pagineInternational Nosology Heritable Disorders of Connective Tissue, Berlin, 1986Willy AndersonNessuna valutazione finora
- Spectrum Cloacal Exstrophy 2011Documento6 pagineSpectrum Cloacal Exstrophy 2011Oscar Sanchez ParisNessuna valutazione finora
- Document on skin changes associated with hypothyroidismDocumento3 pagineDocument on skin changes associated with hypothyroidismKrystal VallesNessuna valutazione finora
- Vitiligo and Leukoderma in Children: Maria Isabel Herane, MDDocumento13 pagineVitiligo and Leukoderma in Children: Maria Isabel Herane, MDBimo Aryo TejoNessuna valutazione finora
- AtherosclerosisDocumento38 pagineAtherosclerosisaffansungkarNessuna valutazione finora
- Vismodegib (Erivedge) For Advanced Basal Cell CarcinomaDocumento7 pagineVismodegib (Erivedge) For Advanced Basal Cell CarcinomaKikin RizkynnisaNessuna valutazione finora
- Abrupt Onset of Dysphagia, Arthritis, Joint Contractures, and Sheets of Monomorphous Waxy PapulesDocumento2 pagineAbrupt Onset of Dysphagia, Arthritis, Joint Contractures, and Sheets of Monomorphous Waxy PapulesRobertoNessuna valutazione finora
- Medip,+2449 8599 1 CEDocumento3 pagineMedip,+2449 8599 1 CEGOD LEVEL LEGEND GAMINGNessuna valutazione finora
- Non Syndromic IcthyosisDocumento109 pagineNon Syndromic IcthyosisSRIRAM CKNessuna valutazione finora
- Widespread Livedoid VasculopathyDocumento4 pagineWidespread Livedoid VasculopathyKata TölgyesiNessuna valutazione finora
- 11 194scleroderma A Case ReportDocumento3 pagine11 194scleroderma A Case ReportAfiatul MukarromahNessuna valutazione finora
- 2019 Zebras in Foreskin Dermatopathology (Natural Circumcision)Documento6 pagine2019 Zebras in Foreskin Dermatopathology (Natural Circumcision)msdsubNessuna valutazione finora
- Congenital Scalp Tumor With Ulceration: Jeremy Udkoff MA, MAS - Ralph E. Holmes MD - Magdalene A. Dohil MDDocumento2 pagineCongenital Scalp Tumor With Ulceration: Jeremy Udkoff MA, MAS - Ralph E. Holmes MD - Magdalene A. Dohil MDAna MariaNessuna valutazione finora
- Scleroderma - Oral Manifestations and Treatment ChallengesDocumento5 pagineScleroderma - Oral Manifestations and Treatment ChallengesLujainNessuna valutazione finora
- Calcinosis Cutis Report of 4 CasesDocumento2 pagineCalcinosis Cutis Report of 4 CasesEtty FaridaNessuna valutazione finora
- Lichen striatus on adultDocumento4 pagineLichen striatus on adultmustafa566512345Nessuna valutazione finora
- Congenital Cutis Laxa Presenting With Hiatal Hernia in The Third Trimester of PregnancyDocumento5 pagineCongenital Cutis Laxa Presenting With Hiatal Hernia in The Third Trimester of PregnancyGabriel SabatkaNessuna valutazione finora
- Skin FinalDocumento3 pagineSkin FinalNikunj PatelNessuna valutazione finora
- Steatocystoma Multiplex Presenting As Acral Subcutaneous NodulesDocumento2 pagineSteatocystoma Multiplex Presenting As Acral Subcutaneous Noduleshendra asusNessuna valutazione finora
- Ichthyosis - Fitzpatrick Dermatology 9th EdDocumento26 pagineIchthyosis - Fitzpatrick Dermatology 9th EdIza Singson-CristobalNessuna valutazione finora
- Keratoacanthoma: A Distinct Entity?: Tobias Gleich, Elena Chiticariu, Marcel Huber and Daniel HohlDocumento7 pagineKeratoacanthoma: A Distinct Entity?: Tobias Gleich, Elena Chiticariu, Marcel Huber and Daniel HohlkikiNessuna valutazione finora
- Skin Signs of Gastrointestinal DiseaseDocumento20 pagineSkin Signs of Gastrointestinal DiseaseMADALINA ENACHENessuna valutazione finora
- Warty Dyskeratoma As A Cutaneous Horn of The Mons PubisDocumento3 pagineWarty Dyskeratoma As A Cutaneous Horn of The Mons PubisDomenica BourneNessuna valutazione finora
- Corneal Vascularization: David G. CoganDocumento9 pagineCorneal Vascularization: David G. CoganPraful ChaudharyNessuna valutazione finora
- HAPP-PICO Tissue Pathology Journal ResearchDocumento4 pagineHAPP-PICO Tissue Pathology Journal ResearchArlynn MartinezNessuna valutazione finora
- Cutaneous Lesions of The Canine ScrotumDocumento14 pagineCutaneous Lesions of The Canine ScrotumLuciana Diegues100% (1)
- Keratoacanthoma of Lip: June 2011Documento3 pagineKeratoacanthoma of Lip: June 2011Gadis PinanditaNessuna valutazione finora
- PIIS019096228 ZDocumento2 paginePIIS019096228 ZSake Cinema21Nessuna valutazione finora
- Patofisiologi Malaria PDFDocumento11 paginePatofisiologi Malaria PDFMeylinda LinNessuna valutazione finora
- Sarcoma de Kaposi PDFDocumento4 pagineSarcoma de Kaposi PDFXavier AltamiranoNessuna valutazione finora
- 2008-Between Light and Dark, The Chimera Comes OutDocumento4 pagine2008-Between Light and Dark, The Chimera Comes OutANessuna valutazione finora
- Parasitología: Experiencia en loxoscelismo cutáneo y cutáneo visceralDocumento10 pagineParasitología: Experiencia en loxoscelismo cutáneo y cutáneo visceralfrank salinasNessuna valutazione finora
- Hers Le 1972Documento4 pagineHers Le 1972Trustia RizqandaruNessuna valutazione finora
- International Wound Journal - 2011 - Percival - Microbiology of The Skin and The Role of Biofilms in InfectionDocumento19 pagineInternational Wound Journal - 2011 - Percival - Microbiology of The Skin and The Role of Biofilms in InfectionTrustia RizqandaruNessuna valutazione finora
- Burkhart 2007Documento3 pagineBurkhart 2007Trustia RizqandaruNessuna valutazione finora
- Kuehnast 2018Documento9 pagineKuehnast 2018Trustia RizqandaruNessuna valutazione finora
- An Increased Incidence of Propionibacterium Acnes Biofilms in Acne Vulgaris: A Case-Control StudyDocumento9 pagineAn Increased Incidence of Propionibacterium Acnes Biofilms in Acne Vulgaris: A Case-Control Studyanon_773807769Nessuna valutazione finora
- Kuehnast 2018Documento9 pagineKuehnast 2018Trustia RizqandaruNessuna valutazione finora
- CoenyeDocumento19 pagineCoenyeTrustia RizqandaruNessuna valutazione finora
- Ab Si Acnee VulgaraDocumento9 pagineAb Si Acnee Vulgaraapotek padasukaNessuna valutazione finora
- Fitz Gibbon2013Documento9 pagineFitz Gibbon2013Trustia RizqandaruNessuna valutazione finora
- From Pathogenesis of Acne Vulgaris To Anti-Acne AgentsDocumento13 pagineFrom Pathogenesis of Acne Vulgaris To Anti-Acne AgentsNazihan Safitri AlkatiriNessuna valutazione finora
- Acne vulgaris pathogenesis and future treatment modalitiesDocumento11 pagineAcne vulgaris pathogenesis and future treatment modalitiesTrustia RizqandaruNessuna valutazione finora
- Cutibacterium Acnes (Propionibacterium Acnes) and Acne Vulgaris: A Brief Look at The Latest UpdatesDocumento10 pagineCutibacterium Acnes (Propionibacterium Acnes) and Acne Vulgaris: A Brief Look at The Latest UpdatesVania Andhika PutriNessuna valutazione finora
- Dos Cutaneus TuberculosisDocumento10 pagineDos Cutaneus TuberculosisTrustia RizqandaruNessuna valutazione finora
- Beluga 3 MobDocumento18 pagineBeluga 3 MobTrustia RizqandaruNessuna valutazione finora
- Biology of MelanocyteDocumento16 pagineBiology of MelanocyteTrustia RizqandaruNessuna valutazione finora
- WilliamsDocumento10 pagineWilliamsTrustia RizqandaruNessuna valutazione finora
- Microbiome in Atopic Dermatitis: Clinical, Cosmetic and Investigational Dermatology DoveDocumento6 pagineMicrobiome in Atopic Dermatitis: Clinical, Cosmetic and Investigational Dermatology DoveTrustia RizqandaruNessuna valutazione finora
- New insights into the structure and barrier functions of skinDocumento15 pagineNew insights into the structure and barrier functions of skinTrustia RizqandaruNessuna valutazione finora
- Desmosome SignalingDocumento24 pagineDesmosome SignalingTrustia RizqandaruNessuna valutazione finora
- 11 Male LUTS PDFDocumento22 pagine11 Male LUTS PDFTrustia RizqandaruNessuna valutazione finora
- DYSLIPIDEMIADocumento61 pagineDYSLIPIDEMIATrustia RizqandaruNessuna valutazione finora
- American College of Rheumatology 2012 Recommendations For The Use of Nonpharmacologic and Pharmacologic Therapies in Osteoarthritis of The Hand, Hip, and KneeDocumento10 pagineAmerican College of Rheumatology 2012 Recommendations For The Use of Nonpharmacologic and Pharmacologic Therapies in Osteoarthritis of The Hand, Hip, and KneeAissyiyah Nur An NisaNessuna valutazione finora
- AmiodaroneDocumento7 pagineAmiodaroneTrustia RizqandaruNessuna valutazione finora
- Gota 2012 Part 2Documento15 pagineGota 2012 Part 2Ps JuNe SnoopyNessuna valutazione finora
- Colloid Use For Fluid ResuscitationDocumento4 pagineColloid Use For Fluid ResuscitationTrustia RizqandaruNessuna valutazione finora
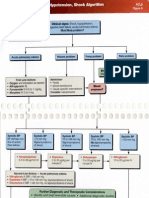
- Acute Pulmonaryl Edema, Hypotension, Shock AlgorithmDocumento2 pagineAcute Pulmonaryl Edema, Hypotension, Shock AlgorithmTrustia RizqandaruNessuna valutazione finora
- Gout Part 1 ACR-12-0014-1Documento16 pagineGout Part 1 ACR-12-0014-1vitauxianaNessuna valutazione finora
- RHDDocumento130 pagineRHDDeepu RajendranNessuna valutazione finora
- Poliomyelitis: Dr. Eiman Sumayyah DPT (Kmu), Ms Neuro (Kmu)Documento14 paginePoliomyelitis: Dr. Eiman Sumayyah DPT (Kmu), Ms Neuro (Kmu)Haniya KhanNessuna valutazione finora
- Pediatrics - Allergy & Immunology TopicsDocumento40 paginePediatrics - Allergy & Immunology TopicspreethamNessuna valutazione finora
- Signs and Symptoms of Common DiseasesDocumento5 pagineSigns and Symptoms of Common DiseasesPeopleNessuna valutazione finora
- Coronary Artery Disease-Cad OR Ischaemic Heart Disease - IhdDocumento99 pagineCoronary Artery Disease-Cad OR Ischaemic Heart Disease - IhdMwanja MosesNessuna valutazione finora
- The Complete Weight Loss Guide: Brought To You byDocumento6 pagineThe Complete Weight Loss Guide: Brought To You bySam YoussefNessuna valutazione finora
- Blood TransfusionDocumento30 pagineBlood TransfusionnoorgianilestariNessuna valutazione finora
- Insert - Elecsys CEA.07027079500.V5.EnDocumento4 pagineInsert - Elecsys CEA.07027079500.V5.EnVegha NedyaNessuna valutazione finora
- Tanda - Tanda Vital Pada Manusia: Jeannete C. Wulandari, Michael Waas, Alan Maulana, Yosa M. Y. ChandraDocumento4 pagineTanda - Tanda Vital Pada Manusia: Jeannete C. Wulandari, Michael Waas, Alan Maulana, Yosa M. Y. ChandraJeannete Claudia WulandariNessuna valutazione finora
- New Microsoft Office Word DocumentDocumento5 pagineNew Microsoft Office Word DocumentMonirul Islam MilonNessuna valutazione finora
- Evo ProductDocumento58 pagineEvo ProductMohammadfaroq HasanNessuna valutazione finora
- DSM-5 and Neurocognitive DisordersDocumento6 pagineDSM-5 and Neurocognitive DisordersluishelNessuna valutazione finora
- Reye SyndromeDocumento15 pagineReye Syndromezeeky35Nessuna valutazione finora
- Other Dance Form: Cheerdance and Contemporary DanceDocumento45 pagineOther Dance Form: Cheerdance and Contemporary DanceJun-jun TulinNessuna valutazione finora
- COPD AE (Book) (20210118) (林冠霖)Documento15 pagineCOPD AE (Book) (20210118) (林冠霖)林冠霖Nessuna valutazione finora
- Dynamicsofdiseasetransmission 160905060144Documento33 pagineDynamicsofdiseasetransmission 160905060144Samjaisheel SamsonNessuna valutazione finora
- Anaesthesia For BurnsDocumento51 pagineAnaesthesia For BurnsAnulatkNessuna valutazione finora
- Hormonal TherapiesDocumento39 pagineHormonal TherapiesJalal EltabibNessuna valutazione finora
- Dialog AssignmentDocumento4 pagineDialog AssignmentKitket Photografi & Cinematografi100% (1)
- Torticolis OdtDocumento25 pagineTorticolis OdtAndreea CimpoiNessuna valutazione finora
- International Travelers Medical Center PrescriptionDocumento1 paginaInternational Travelers Medical Center PrescriptionDeo Warren SilangNessuna valutazione finora
- RANZCO 2012 PosterDocumento7 pagineRANZCO 2012 PosterGray Design GroupNessuna valutazione finora
- Diagnosis and Treatment of Clostridioides (Clostridium) Difficile Infection in Adults in 2020Documento2 pagineDiagnosis and Treatment of Clostridioides (Clostridium) Difficile Infection in Adults in 2020Alem OrihuelaNessuna valutazione finora
- Immune Disorders..Ppt FairDocumento79 pagineImmune Disorders..Ppt FairSumi SebastianNessuna valutazione finora
- BSC Nursing: Medical Surgical Nursing - I Unit V - Disorders of The Cardio Vascular SystemDocumento36 pagineBSC Nursing: Medical Surgical Nursing - I Unit V - Disorders of The Cardio Vascular SystemPoova RagavanNessuna valutazione finora
- 3021 Reflective JournalDocumento5 pagine3021 Reflective Journalapi-660581645Nessuna valutazione finora
- Vit DDocumento36 pagineVit DConstantin MarioaraNessuna valutazione finora
- Multiple MyelomaDocumento8 pagineMultiple MyelomaFelipeNessuna valutazione finora
- Cerebrospinal Fluid (CSF) Sample RequirementsDocumento7 pagineCerebrospinal Fluid (CSF) Sample RequirementsDr.Nouf alhasawiNessuna valutazione finora
- Thromboembolic Disease in Pregnancy د.علية شعيبDocumento50 pagineThromboembolic Disease in Pregnancy د.علية شعيبMohammad Belbahaith0% (1)
- Diabetic Ketoacidosis CQBDocumento9 pagineDiabetic Ketoacidosis CQBkajanyloganNessuna valutazione finora