Potrebbero piacerti anche
- Chapter 7 - Energy Balance For Closed and Open Systems - AzizulDocumento102 pagineChapter 7 - Energy Balance For Closed and Open Systems - Azizulmuhammad izzulNessuna valutazione finora
- ThermoIIIDocumento45 pagineThermoIIInisa hafifahNessuna valutazione finora
- Chap 3 ThermochemistryDocumento51 pagineChap 3 ThermochemistryChong RuYinNessuna valutazione finora
- Study Unit 2 First Law BBDocumento47 pagineStudy Unit 2 First Law BBLutendo Assurance MadzivhaaNessuna valutazione finora
- Energy ConceptsDocumento20 pagineEnergy Conceptscarlos peñaNessuna valutazione finora
- First Law Thermo PDFDocumento45 pagineFirst Law Thermo PDFIbrahim AliNessuna valutazione finora
- Energy Changes in Chemical ReactionsDocumento32 pagineEnergy Changes in Chemical ReactionsRon allen ConconNessuna valutazione finora
- The First Law and Other Basic Concepts (Part 1)Documento23 pagineThe First Law and Other Basic Concepts (Part 1)Ragil BudiartoNessuna valutazione finora
- Week 7 2014Documento21 pagineWeek 7 2014Nicole Anne BorromeoNessuna valutazione finora
- 7 2 PDFDocumento103 pagine7 2 PDFmuhammad izzulNessuna valutazione finora
- Study ChemDocumento13 pagineStudy ChemJanthina Rose AusteroNessuna valutazione finora
- Aula 6Documento57 pagineAula 6JehuNessuna valutazione finora
- Cursul 1 Termodinamica 1 Merged CompressedDocumento182 pagineCursul 1 Termodinamica 1 Merged CompressedTeodor OlaruNessuna valutazione finora
- 002general Energy Analysis of THERMODYNAMICSDocumento30 pagine002general Energy Analysis of THERMODYNAMICSm7sen mohammedNessuna valutazione finora
- Chemistry 2 Module 3Documento10 pagineChemistry 2 Module 3Joshua James Sanguenza RodriguezNessuna valutazione finora
- CHM432 Chap 1Documento57 pagineCHM432 Chap 1KhalijahNessuna valutazione finora
- Chapter - 2 For e ClassDocumento25 pagineChapter - 2 For e Classparsa.shakeri.bavilNessuna valutazione finora
- Thermodyamic Presentation PDFDocumento15 pagineThermodyamic Presentation PDFsilvermist235Nessuna valutazione finora
- Chapter 1 ThermochemistryDocumento78 pagineChapter 1 Thermochemistrymikki11Nessuna valutazione finora
- Energy Balance SKDocumento38 pagineEnergy Balance SKMegaraj ReddyNessuna valutazione finora
- ThermochemistryDocumento50 pagineThermochemistrythobyy100% (4)
- Engineering Mechanics: Second PartDocumento18 pagineEngineering Mechanics: Second Partاحمد سلمان عزيز , مسائيCNessuna valutazione finora
- Lecture 3 ThermodynamicsDocumento54 pagineLecture 3 Thermodynamicstorawe6575Nessuna valutazione finora
- The First Law: Physical ChemistryDocumento26 pagineThe First Law: Physical ChemistryAnnaLynYepesNessuna valutazione finora
- L2 (Energy Analysis)Documento31 pagineL2 (Energy Analysis)Kavin KabilanNessuna valutazione finora
- Thermo Chemistry-01-TheoryDocumento15 pagineThermo Chemistry-01-TheoryRaju SinghNessuna valutazione finora
- General Chemistry L5Documento25 pagineGeneral Chemistry L5Ghassan AteelyNessuna valutazione finora
- Energetics of Chemical ReactionsDocumento58 pagineEnergetics of Chemical ReactionsBichitra GautamNessuna valutazione finora
- Lecture-17 & 18Documento8 pagineLecture-17 & 18Dedar RashidNessuna valutazione finora
- Chapter 03 Thermodynamics PDFDocumento101 pagineChapter 03 Thermodynamics PDFPutri Nur Aisyah Halmy AzamNessuna valutazione finora
- CET I 2.first Law 2021Documento40 pagineCET I 2.first Law 2021Dhruv AgarwalNessuna valutazione finora
- Topic+2 +1st+Law+Thermodynamics+1+Aug+2022Documento40 pagineTopic+2 +1st+Law+Thermodynamics+1+Aug+2022mpumelaqqNessuna valutazione finora
- G-11 Physics U-5 NoteDocumento30 pagineG-11 Physics U-5 Noteaffiliate1326Nessuna valutazione finora
- Thermochemistry NotesDocumento5 pagineThermochemistry NotesNephtali Pinos-anNessuna valutazione finora
- 1-Chapter Four Chemical Dynamics-2016-First PartDocumento42 pagine1-Chapter Four Chemical Dynamics-2016-First PartMOHAMMAD NAZMUL ISLAMNessuna valutazione finora
- C1 ThermochemistryDocumento39 pagineC1 ThermochemistryaliesyaNessuna valutazione finora
- E233 - Thermofluids: The First Law of ThermodynamicsDocumento12 pagineE233 - Thermofluids: The First Law of ThermodynamicsYingyote LubphooNessuna valutazione finora
- ThermodynamicsDocumento82 pagineThermodynamicsmdnishathasan141Nessuna valutazione finora
- Engineering ThermodynamicsDocumento60 pagineEngineering ThermodynamicsJeyaram KumarNessuna valutazione finora
- Lecture Handouts-2 2Documento35 pagineLecture Handouts-2 2Ibrahim HersiNessuna valutazione finora
- LESSON 3 Chemistry FuelDocumento11 pagineLESSON 3 Chemistry Fuelsimonjohn spanglerNessuna valutazione finora
- Ch06 SlidesDocumento73 pagineCh06 SlidesbeelzeburtonNessuna valutazione finora
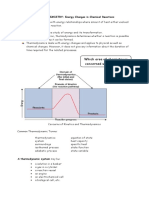
- Which Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsDocumento19 pagineWhich Area of Chemistry Is Concerned With The Topic?: THERMOCHEMISTRY: Energy Changes in Chemical ReactionsSahada KanapiyaNessuna valutazione finora
- By Rachel & AmitDocumento23 pagineBy Rachel & AmitAmit ShahNessuna valutazione finora
- Chap 2 BDocumento18 pagineChap 2 BMike BelayNessuna valutazione finora
- 2-Energy LawsDocumento4 pagine2-Energy LawsBlue HeavensNessuna valutazione finora
- Physical Chemistry (Part-2)Documento73 paginePhysical Chemistry (Part-2)RSLNessuna valutazione finora
- CP Ch6Documento30 pagineCP Ch6zgazga amirNessuna valutazione finora
- 1.1.3 The First Law of ThermodynamicsDocumento4 pagine1.1.3 The First Law of ThermodynamicsRomeo San GasparNessuna valutazione finora
- ThermochemistryDocumento12 pagineThermochemistrynsuperticiosoNessuna valutazione finora
- Module 3Documento11 pagineModule 3Teofilo Matthew AriñoNessuna valutazione finora
- Lesson 3: The First Law of Thermodynamics: Module 1: Energy Chapter 1: FuelsDocumento4 pagineLesson 3: The First Law of Thermodynamics: Module 1: Energy Chapter 1: Fuelscory kurdapyaNessuna valutazione finora
- ch1 PDFDocumento26 paginech1 PDFDono SusilNessuna valutazione finora
- P 7.4 ThermochemistryDocumento32 pagineP 7.4 ThermochemistryFelicia GunawanNessuna valutazione finora
- Laws of Thermodyanmics PDFDocumento33 pagineLaws of Thermodyanmics PDFAljohn Mark ReyesNessuna valutazione finora
- Enthalpy 1Documento102 pagineEnthalpy 1kobegwapo595Nessuna valutazione finora
- CCTD101B Notes 4 - 1st Law of ThermodynamicsDocumento7 pagineCCTD101B Notes 4 - 1st Law of ThermodynamicsShan ShanzNessuna valutazione finora
- Energy and The First Law of Thermodynamics: Prepared By: EFREN A. DELA CRUZ E-Mail Address: Eadelacruz@clsu - Edu.phDocumento7 pagineEnergy and The First Law of Thermodynamics: Prepared By: EFREN A. DELA CRUZ E-Mail Address: Eadelacruz@clsu - Edu.phBilly Jake CorpuzNessuna valutazione finora
- QuestionsDocumento8 pagineQuestionsAntonioNessuna valutazione finora
- “Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4Da Everand“Foundations to Flight: Mastering Physics from Curiosity to Confidence: Cipher 4”: “Foundations to Flight: Mastering Physics from Curiosity to Confidence, #4Nessuna valutazione finora
- PH100 Tutorial 02Documento3 paginePH100 Tutorial 02Yash SinghNessuna valutazione finora
- ThermoDocumento12 pagineThermokesiled309Nessuna valutazione finora
- AP Physics 2Documento155 pagineAP Physics 2Faiyaaz KarimNessuna valutazione finora
- Basic Physics Notes by Atul WaghmareDocumento46 pagineBasic Physics Notes by Atul WaghmareAtulWaghmareNessuna valutazione finora
- RefrigerationDocumento163 pagineRefrigerationali105Nessuna valutazione finora
- Brainkart 1NQKLHuNY5E 83rhvrbqCeQMF4f5KTkGz - pdf-1Documento16 pagineBrainkart 1NQKLHuNY5E 83rhvrbqCeQMF4f5KTkGz - pdf-1Revanth RamineniNessuna valutazione finora
- Large Scale Compressed Hydrogen StorageDocumento20 pagineLarge Scale Compressed Hydrogen Storagepec21102002100% (1)
- ThermodynamicsDocumento13 pagineThermodynamicsabhishekNessuna valutazione finora
- Heat and Thermodynamics According To KPK Textbook & Sindh TextbookDocumento15 pagineHeat and Thermodynamics According To KPK Textbook & Sindh Textbookswatmiandam44Nessuna valutazione finora
- ECH148 UC DavisDocumento5 pagineECH148 UC DavisKaul PatrickNessuna valutazione finora
- Thermodynamics PDFDocumento202 pagineThermodynamics PDFLawNessuna valutazione finora
- Thermodynamics MCQDocumento13 pagineThermodynamics MCQBhaskar jyoti BhuyanNessuna valutazione finora
- Thermodynamics MST 2011Documento3 pagineThermodynamics MST 2011greenhoochyNessuna valutazione finora
- Exercicios Ch06 FundamentalsEngineeringThermodynamics7e PDFDocumento23 pagineExercicios Ch06 FundamentalsEngineeringThermodynamics7e PDFAndri YansyahNessuna valutazione finora
- Practice Problems ThermodynamicsDocumento5 paginePractice Problems ThermodynamicsJana ChambersNessuna valutazione finora
- Lecture 4 - Steam CyclesDocumento20 pagineLecture 4 - Steam CyclesWillie MojataleNessuna valutazione finora
- Indiabix ThermoDocumento44 pagineIndiabix ThermoZir AeNessuna valutazione finora
- 4.thermodynamics and Thermochemistry TheoryDocumento39 pagine4.thermodynamics and Thermochemistry TheoryRithvikNessuna valutazione finora
- 2020 2021 Intake CHE 450 ASSGN 2Documento3 pagine2020 2021 Intake CHE 450 ASSGN 2Lad SlasNessuna valutazione finora
- Thermodynamic 1Documento312 pagineThermodynamic 1Daniel Wang100% (5)
- Power Estimation For Air Cooling and Dehumidification Using Exergy AnalysisDocumento11 paginePower Estimation For Air Cooling and Dehumidification Using Exergy AnalysisAlexander VovaNessuna valutazione finora
- Problems 21 Serway 6 eDocumento12 pagineProblems 21 Serway 6 eOmari McDuffeyNessuna valutazione finora
- JEE Main 2024 January 27 Questions With Solutions by Reli Odi66BCDocumento33 pagineJEE Main 2024 January 27 Questions With Solutions by Reli Odi66BCHarikesh SharmaNessuna valutazione finora
- CEIC2000 Thermodynamics - Lecture 2 The First Law Forms of EnergyDocumento49 pagineCEIC2000 Thermodynamics - Lecture 2 The First Law Forms of EnergyJoshua JohnNessuna valutazione finora
- Chapter # 11 HeatDocumento6 pagineChapter # 11 HeatSIR USMAN KHAN50% (2)
- Thermal and Power Plant Engineering2 PDFDocumento36 pagineThermal and Power Plant Engineering2 PDFChaitanya Kishore ChitikenaNessuna valutazione finora
- Second Law of ThermodynamicsDocumento21 pagineSecond Law of ThermodynamicsVaibhav Vithoba NaikNessuna valutazione finora
- Course Objective QuestionDocumento243 pagineCourse Objective Questionahmish kabbaxeNessuna valutazione finora
- An Introduction To Stirling-Cycle MachinesDocumento11 pagineAn Introduction To Stirling-Cycle MachinessandigricNessuna valutazione finora
- Bme Question Bank PDFDocumento41 pagineBme Question Bank PDFnagarajan224Nessuna valutazione finora