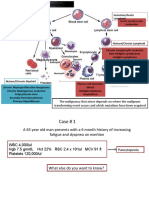
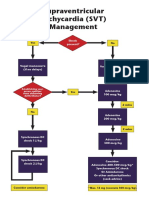
Potrebbero piacerti anche
- Cwu 1 OrthoDocumento14 pagineCwu 1 OrthoHakimah K. Suhaimi100% (1)
- Understanding PancytopeniaDocumento68 pagineUnderstanding PancytopeniaThaveeshaLindsayWhiteNessuna valutazione finora
- MDSDocumento46 pagineMDSFesti Mada HelmiNessuna valutazione finora
- Psycho Dynamic Psychotherapy For Personality DisordersDocumento40 paginePsycho Dynamic Psychotherapy For Personality DisorderslhasniaNessuna valutazione finora
- Herbal Toothpaste FactsDocumento26 pagineHerbal Toothpaste Factsmahek_dhootNessuna valutazione finora
- Eye Ear Dental Examinations Form 3300Documento2 pagineEye Ear Dental Examinations Form 3300Khyaree EspañaNessuna valutazione finora
- FC Paed (SA) Part I Past Papers - 2011 Sept 6-4-2014Documento8 pagineFC Paed (SA) Part I Past Papers - 2011 Sept 6-4-2014matenten0% (1)
- Mullally - ASH Hematology Review Series - MPN - CombinedDocumento40 pagineMullally - ASH Hematology Review Series - MPN - CombinedИван НегарэNessuna valutazione finora
- MDS MPN2019 DR Rachel SalitDocumento39 pagineMDS MPN2019 DR Rachel Salitluca.win92Nessuna valutazione finora
- Acute Leukaemia-Update: DR Niranjan N. RathodDocumento89 pagineAcute Leukaemia-Update: DR Niranjan N. RathodratanNessuna valutazione finora
- Medicine2 - Myeloproliferative, Lymphoproliferative WorkshopDocumento118 pagineMedicine2 - Myeloproliferative, Lymphoproliferative Workshopapi-3762917100% (1)
- Clinical approach to bleeding problems in childhoodDocumento38 pagineClinical approach to bleeding problems in childhoodbennyrolandnababanNessuna valutazione finora
- How I Treat Essential ThrombocythemiaDocumento12 pagineHow I Treat Essential ThrombocythemiaJicko Street HooligansNessuna valutazione finora
- Acute and Chronic Leukemia FinalDocumento68 pagineAcute and Chronic Leukemia FinalHannah LeiNessuna valutazione finora
- Heme Malignancy Review 2020Documento163 pagineHeme Malignancy Review 2020Anna StacyNessuna valutazione finora
- LeukemiaDocumento91 pagineLeukemiaShadin SNessuna valutazione finora
- Hema Platelet DisordersDocumento13 pagineHema Platelet DisordersJennie Grace MaloomNessuna valutazione finora
- Chronic Leukemia Lecture.Documento47 pagineChronic Leukemia Lecture.sherifref3atNessuna valutazione finora
- Kemia: Reported byDocumento49 pagineKemia: Reported bykris_ocio963Nessuna valutazione finora
- Mengenal Kanker Darah2Documento24 pagineMengenal Kanker Darah2juwitaNessuna valutazione finora
- Handout 2 1522436248 PDFDocumento82 pagineHandout 2 1522436248 PDFHarnadi WonogiriNessuna valutazione finora
- Aspek Laboratorium BCR Abl Kuantitatif & JAK2 V617FDocumento76 pagineAspek Laboratorium BCR Abl Kuantitatif & JAK2 V617FullifannuriNessuna valutazione finora
- Clinical Aspect & Therapy of Acute LeukemiaDocumento28 pagineClinical Aspect & Therapy of Acute LeukemiaImtihanamiseNessuna valutazione finora
- Leukemia 2Documento52 pagineLeukemia 2Luthfia WardhaniNessuna valutazione finora
- 11-23-21 White Blood Cell DisordersDocumento80 pagine11-23-21 White Blood Cell DisordersdeNessuna valutazione finora
- Keganasan HematologiDocumento69 pagineKeganasan HematologiRuki HartawanNessuna valutazione finora
- LeukemiaDocumento40 pagineLeukemiaYeni Jesika SilitongaNessuna valutazione finora
- AML Pita DR MardiahDocumento71 pagineAML Pita DR MardiahSarly Puspita AriesaNessuna valutazione finora
- Chronic Leukaemia 111Documento15 pagineChronic Leukaemia 111Razib HasanNessuna valutazione finora
- ThrombocytopeniaDocumento49 pagineThrombocytopeniaMaria EnjelinaNessuna valutazione finora
- Myeloproliferative Disorders: Classification CML AMM PV ETDocumento58 pagineMyeloproliferative Disorders: Classification CML AMM PV ETashuNessuna valutazione finora
- Lecture SlidesDocumento86 pagineLecture SlidesJyotsna NigamNessuna valutazione finora
- Acute Leukemia Diagnosis, Treatment, PrognosisDocumento31 pagineAcute Leukemia Diagnosis, Treatment, PrognosisfranzzjosefNessuna valutazione finora
- Understanding Acute and Chronic LeukemiaDocumento10 pagineUnderstanding Acute and Chronic LeukemiaKrisha BalorioNessuna valutazione finora
- Acute Leukemia in ChildrenDocumento15 pagineAcute Leukemia in ChildrenSabrina JonesNessuna valutazione finora
- WhddidsorhkDocumento50 pagineWhddidsorhkestherin909Nessuna valutazione finora
- OPCIONAL - Myeloproliferative NeoplasmsDocumento44 pagineOPCIONAL - Myeloproliferative NeoplasmsGabriel CohenNessuna valutazione finora
- LeukemiaDocumento51 pagineLeukemiaKailash KhatriNessuna valutazione finora
- Primary and Secondary Thrombocytosis in ChildhoodDocumento13 paginePrimary and Secondary Thrombocytosis in ChildhoodSofia RuanoNessuna valutazione finora
- Hematolocical Clonal Disorders (Malignancies) : Lympho-Proliferative Disorders Myeloid DisordersDocumento50 pagineHematolocical Clonal Disorders (Malignancies) : Lympho-Proliferative Disorders Myeloid DisordersAngelica CoppolaNessuna valutazione finora
- Teruko Sakurai Japanese Color Harmony DictionaryDocumento22 pagineTeruko Sakurai Japanese Color Harmony DictionaryLily AddamsNessuna valutazione finora
- MYELODYSPLASTIC SYNDROME AND MULTIPLE MYELOMADocumento46 pagineMYELODYSPLASTIC SYNDROME AND MULTIPLE MYELOMAdelaNessuna valutazione finora
- Myeloproliferative Disorders (Bhs Inggris)Documento57 pagineMyeloproliferative Disorders (Bhs Inggris)Denny DedenNessuna valutazione finora
- Myeloproliferative DisordersDocumento2 pagineMyeloproliferative DisordersGerardLumNessuna valutazione finora
- Acute Myeloid Leukemia - AML: Clinical BackgroundDocumento12 pagineAcute Myeloid Leukemia - AML: Clinical BackgroundNyxa AbdullaNessuna valutazione finora
- Chronic Myeloid Leukemia (CML) - Hematology and Oncology - MSD Manual Professional EditionDocumento3 pagineChronic Myeloid Leukemia (CML) - Hematology and Oncology - MSD Manual Professional Editionnurul auliaNessuna valutazione finora
- Hematology Review 2021-2Documento142 pagineHematology Review 2021-2Maram AbdullahNessuna valutazione finora
- Myeloproliferative DisorderDocumento36 pagineMyeloproliferative DisorderKalpana ShahNessuna valutazione finora
- Leukemia 16-12-2019vDocumento38 pagineLeukemia 16-12-2019vChrisandiNessuna valutazione finora
- Pathology of Blood MalignancyDocumento38 paginePathology of Blood MalignancyHappy FluffyNessuna valutazione finora
- The LeukemiasDocumento52 pagineThe Leukemiasمصطفي خندقاوي100% (1)
- Cancers 12 01746Documento18 pagineCancers 12 01746LauraQuinteroNessuna valutazione finora
- Cancers 12 01746 v2Documento18 pagineCancers 12 01746 v2Ulicer CruzNessuna valutazione finora
- 5 Myeloproliferative NeoplasmsDocumento49 pagine5 Myeloproliferative NeoplasmsEliza Stanescu100% (1)
- Anemia Lectures From Doll Very Well Covered in Pathoma Coberly Plasma Cell DyscrasiasDocumento19 pagineAnemia Lectures From Doll Very Well Covered in Pathoma Coberly Plasma Cell DyscrasiasTeehee JonesNessuna valutazione finora
- ACUTE LYMPHOBLASTIC LEUKEMIA (ALLDocumento12 pagineACUTE LYMPHOBLASTIC LEUKEMIA (ALLsharon victoria mendezNessuna valutazione finora
- Kuliah 14.3 - Chronic Myeloid Leukemia (Dr. Ali Santosa)Documento32 pagineKuliah 14.3 - Chronic Myeloid Leukemia (Dr. Ali Santosa)Fikri UdinNessuna valutazione finora
- Acute Lymphoid Leukemia (Airway Management)Documento48 pagineAcute Lymphoid Leukemia (Airway Management)Ankita SamantaNessuna valutazione finora
- Laboratory Evaluation of Hemostatic and Coagulation AbnormalitiesDocumento68 pagineLaboratory Evaluation of Hemostatic and Coagulation Abnormalitiesivana.begic.1960Nessuna valutazione finora
- Normal Hematolymphoid TissuesDocumento182 pagineNormal Hematolymphoid TissuescandiddreamsNessuna valutazione finora
- Interpretation of CBC: Dr. N. BajajDocumento56 pagineInterpretation of CBC: Dr. N. BajajHaSan Z. MustafaNessuna valutazione finora
- Acute LeukaemiaDocumento39 pagineAcute LeukaemiaAbby LiewNessuna valutazione finora
- Diseases of The Bone Marrow and Blood Conditions That Can Occur When The Blood-Forming Cells in The Bone Marrow Become AbnormalDocumento4 pagineDiseases of The Bone Marrow and Blood Conditions That Can Occur When The Blood-Forming Cells in The Bone Marrow Become AbnormalLuqman QadirNessuna valutazione finora
- Enteral Vs Parenteral, Treatment Option For Hipoalbumin Management in IcuDocumento34 pagineEnteral Vs Parenteral, Treatment Option For Hipoalbumin Management in IcusiputleletNessuna valutazione finora
- Daftar Keg Ilmiah 2017 PDFDocumento52 pagineDaftar Keg Ilmiah 2017 PDFFAEZYA DEWI100% (1)
- Efficacy of Short Course PDFDocumento14 pagineEfficacy of Short Course PDFsiputleletNessuna valutazione finora
- S1.1 Ria Bandiara - Role Managemen Hypertension PKB 2019Documento29 pagineS1.1 Ria Bandiara - Role Managemen Hypertension PKB 2019siputleletNessuna valutazione finora
- Kars 2017 - 2Documento34 pagineKars 2017 - 2siputleletNessuna valutazione finora
- WS16 Uun S - Empiric Antimicrobial Management of SepsisDocumento12 pagineWS16 Uun S - Empiric Antimicrobial Management of SepsissiputleletNessuna valutazione finora
- Heart Failure Management Pathway: What Is New?Documento50 pagineHeart Failure Management Pathway: What Is New?siputleletNessuna valutazione finora
- Ethicolegal Aspects of Medical PracticeDocumento31 pagineEthicolegal Aspects of Medical PracticesiputleletNessuna valutazione finora
- LS1.2 Rudi W - Dengue Management and Prevention PKB 2019Documento27 pagineLS1.2 Rudi W - Dengue Management and Prevention PKB 2019DhannyNessuna valutazione finora
- Efficacy of Short Course PDFDocumento14 pagineEfficacy of Short Course PDFsiputleletNessuna valutazione finora
- WS16 Uun S - Empiric Antimicrobial Management of SepsisDocumento12 pagineWS16 Uun S - Empiric Antimicrobial Management of SepsissiputleletNessuna valutazione finora
- Bcu 2018 Final Announcement-RevDocumento16 pagineBcu 2018 Final Announcement-RevsiputleletNessuna valutazione finora
- Dr. Laniyati Hamijoyo Sppd-Kr. M.Kes: - Staf Pengajar Divisi Reumatologi Departemen IlmuDocumento71 pagineDr. Laniyati Hamijoyo Sppd-Kr. M.Kes: - Staf Pengajar Divisi Reumatologi Departemen IlmusiputleletNessuna valutazione finora
- E-Ticket: Departure FlightDocumento2 pagineE-Ticket: Departure FlightsiputleletNessuna valutazione finora
- Macam-Macam Bencana & Risiko Kerawanan Di Indonesia: Aryono D.Pusponegoro Yayasan AGD 118Documento32 pagineMacam-Macam Bencana & Risiko Kerawanan Di Indonesia: Aryono D.Pusponegoro Yayasan AGD 118siputleletNessuna valutazione finora
- 2nd Announcement APSCCEM 2019Documento20 pagine2nd Announcement APSCCEM 2019siputleletNessuna valutazione finora
- PCRA Review Construction RisksDocumento11 paginePCRA Review Construction RiskssiputleletNessuna valutazione finora
- 2nd Sunshine 2018 EdDocumento2 pagine2nd Sunshine 2018 EdsiputleletNessuna valutazione finora
- Algorithms - SVT PDFDocumento1 paginaAlgorithms - SVT PDFjhontarroNessuna valutazione finora
- Efficacy of Short Course PDFDocumento14 pagineEfficacy of Short Course PDFsiputleletNessuna valutazione finora
- Daftar Keg Ilmiah 2017 PDFDocumento52 pagineDaftar Keg Ilmiah 2017 PDFFAEZYA DEWI100% (1)
- Efficacy of Short Course PDFDocumento14 pagineEfficacy of Short Course PDFsiputleletNessuna valutazione finora
- Declaration of Erin EllisDocumento4 pagineDeclaration of Erin EllisElizabeth Nolan BrownNessuna valutazione finora
- The Effectiveness of Community-Based Rehabilitation Programs For Person Who Use Drugs (PWUD) : Perspectives of Rehabilitation Care Workers in IloiloDocumento27 pagineThe Effectiveness of Community-Based Rehabilitation Programs For Person Who Use Drugs (PWUD) : Perspectives of Rehabilitation Care Workers in IloiloErikah Eirah BeloriaNessuna valutazione finora
- Research Paper 4Documento26 pagineResearch Paper 4Amit RajputNessuna valutazione finora
- Sspc-Ab 2Documento3 pagineSspc-Ab 2HafidzManafNessuna valutazione finora
- BICEP GROWTHDocumento3 pagineBICEP GROWTHJee MusaNessuna valutazione finora
- "We Are Their Slaves" by Gregory FlanneryDocumento4 pagine"We Are Their Slaves" by Gregory FlanneryAndy100% (2)
- C & W Meat Packers Carly Courtney Cynthiana KY Notice of Suspension Humane Treatment LivestockDocumento4 pagineC & W Meat Packers Carly Courtney Cynthiana KY Notice of Suspension Humane Treatment LivestockghostgripNessuna valutazione finora
- Sabrina Nelson Winters Op Ed - BHM 2024Documento2 pagineSabrina Nelson Winters Op Ed - BHM 2024Andrew WilsonNessuna valutazione finora
- Anand - 1994 - Fluorouracil CardiotoxicityDocumento5 pagineAnand - 1994 - Fluorouracil Cardiotoxicityaly alyNessuna valutazione finora
- Improving Healthcare Quality in IndonesiaDocumento34 pagineImproving Healthcare Quality in IndonesiaJuanaNessuna valutazione finora
- Abc Sealant SDSDocumento5 pagineAbc Sealant SDSKissa DolautaNessuna valutazione finora
- Dr. Banis' Psychosomatic Energetics Method for Healing Health ConditionsDocumento34 pagineDr. Banis' Psychosomatic Energetics Method for Healing Health ConditionsmhadarNessuna valutazione finora
- Ophthalmology 7th Edition (Oftalmología 7a Edición)Documento1.671 pagineOphthalmology 7th Edition (Oftalmología 7a Edición)Víctor SaRi100% (1)
- Csa Fodrea 2014 - 2015 Student Handbook FinalDocumento37 pagineCsa Fodrea 2014 - 2015 Student Handbook Finalapi-260407035Nessuna valutazione finora
- Hortatory Exposition Humaira AssahdaDocumento4 pagineHortatory Exposition Humaira Assahdaaleeka auroraNessuna valutazione finora
- Low Back Pain Dr. Hardhi PRanataDocumento57 pagineLow Back Pain Dr. Hardhi PRanataPerwita ArumingtyasNessuna valutazione finora
- New TNMDocumento157 pagineNew TNMShouvik ChowdhuryNessuna valutazione finora
- MadRiverUnion01 13 21editionDocumento8 pagineMadRiverUnion01 13 21editionMad River UnionNessuna valutazione finora
- Marriage and Later PartDocumento25 pagineMarriage and Later PartDeepak PoudelNessuna valutazione finora
- PE 11 Cardio Endurance Workout ReflectionsDocumento2 paginePE 11 Cardio Endurance Workout ReflectionsFranky MendozaNessuna valutazione finora
- Tugas English Geri PebriansyahDocumento10 pagineTugas English Geri PebriansyahAyu AndiniiNessuna valutazione finora
- 2018 Intervention Session Template 3Documento4 pagine2018 Intervention Session Template 3api-404544260Nessuna valutazione finora
- Corn SpecDocumento4 pagineCorn SpecJohanna MullerNessuna valutazione finora
- Atwwi "Virtual" Trading Room Reference Document November 2, 2020Documento4 pagineAtwwi "Virtual" Trading Room Reference Document November 2, 2020amisamiam2Nessuna valutazione finora
- The Child Bipolar QuestionnaireDocumento10 pagineThe Child Bipolar QuestionnairefranciscatomiNessuna valutazione finora