Potrebbero piacerti anche
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersDa EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNessuna valutazione finora
- Validation Master PlanDocumento29 pagineValidation Master Planspark80988100% (6)
- Practical Approaches to Method Validation and Essential Instrument QualificationDa EverandPractical Approaches to Method Validation and Essential Instrument QualificationNessuna valutazione finora
- VAL 080 Validation Master Plan Sample PDFDocumento3 pagineVAL 080 Validation Master Plan Sample PDFsiva sankar100% (1)
- Tim Fields Master Validation PlanDocumento7 pagineTim Fields Master Validation Planmanoj262400/2100% (1)
- Process Validation A Complete Guide - 2020 EditionDa EverandProcess Validation A Complete Guide - 2020 EditionNessuna valutazione finora
- SOP - 0400 - 10 - Design Qualification SOPDocumento13 pagineSOP - 0400 - 10 - Design Qualification SOPsandipkumardshahNessuna valutazione finora
- Risk Based Validation and Requalification of Processes EquipmentDocumento33 pagineRisk Based Validation and Requalification of Processes EquipmentfenixseravgonNessuna valutazione finora
- Validation Master PlanDocumento5 pagineValidation Master Planazamyn83% (6)
- 2015 VMP TemplateDocumento10 pagine2015 VMP Templatekulbhushan singh100% (2)
- Validation Protocol CG TADocumento30 pagineValidation Protocol CG TACarlo Duran100% (1)
- Cleaning ValidationDocumento21 pagineCleaning ValidationBrian WilliamsNessuna valutazione finora
- SOP On Handling of DeviationsDocumento9 pagineSOP On Handling of DeviationsBlueSaga100% (1)
- SOP Equipment ValidationDocumento15 pagineSOP Equipment Validationfarjana100% (7)
- Validation ProtocolDocumento63 pagineValidation ProtocolIndústria Petys64% (22)
- Validation Master Plan TemplateDocumento17 pagineValidation Master Plan TemplateNadine100% (4)
- Cleaning Validation ProtocolDocumento3 pagineCleaning Validation Protocolpuneetogupta100% (1)
- VMP For EVFDocumento56 pagineVMP For EVFPrashansa ShresthaNessuna valutazione finora
- Laboratory Quality Agreement TamplateDocumento10 pagineLaboratory Quality Agreement TamplateMina Maher MikhailNessuna valutazione finora
- Site VMPDocumento34 pagineSite VMPJonatan Dominguez PerezNessuna valutazione finora
- VMP 01Documento25 pagineVMP 01Atul Sharma100% (2)
- U R T For A Supervisory Control and Data Acquisition (SCADA) Process Control SystemDocumento65 pagineU R T For A Supervisory Control and Data Acquisition (SCADA) Process Control Systemkapernikov50% (2)
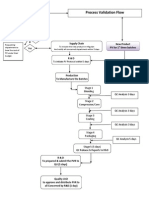
- Flow Chart Process ValidationDocumento1 paginaFlow Chart Process Validationmasthan6yNessuna valutazione finora
- Validation VialWasher OQ NIHDocumento30 pagineValidation VialWasher OQ NIHcongacon3aNessuna valutazione finora
- ISPE ChecklistDocumento6 pagineISPE Checklistm_ihab777629Nessuna valutazione finora
- Eu GMPDocumento16 pagineEu GMPamirin_kingNessuna valutazione finora
- HVAC QualificationDocumento36 pagineHVAC QualificationMuhammadAteeq100% (3)
- CIQA Validation Master Plan Sample TemplateDocumento4 pagineCIQA Validation Master Plan Sample TemplateSatyam Gupta100% (1)
- Validation PolicyDocumento3 pagineValidation PolicyneppoanandNessuna valutazione finora
- Iq Oq PQ ChamberDocumento17 pagineIq Oq PQ Chamberelias_7760% (5)
- Validation VMP Validation Master PlanDocumento13 pagineValidation VMP Validation Master Plank.p.Nessuna valutazione finora
- Facility ValidationDocumento12 pagineFacility ValidationGhanta Ranjith Kumar100% (1)
- Validation Master PlanDocumento37 pagineValidation Master PlanOsama Mahmoud0% (1)
- Validation and Facility Design PDFDocumento16 pagineValidation and Facility Design PDFjpabloqfNessuna valutazione finora
- Installation Qualification Protocol For Walk - in Incubator System Name: Walk - in Incubator Document No.: NECPL/IQ/11220I/1 Page No.: 1 of 25Documento25 pagineInstallation Qualification Protocol For Walk - in Incubator System Name: Walk - in Incubator Document No.: NECPL/IQ/11220I/1 Page No.: 1 of 25vipin100% (1)
- VAL-085 Process Validation Guideline SampleDocumento2 pagineVAL-085 Process Validation Guideline SampleVizit31Nessuna valutazione finora
- OOS ReportDocumento2 pagineOOS ReportAnton MymrikovNessuna valutazione finora
- SOP - 0700 - 10 - Validation Summary SOPDocumento9 pagineSOP - 0700 - 10 - Validation Summary SOPdharmil.gopaniNessuna valutazione finora
- IQOQ ProtocolDocumento4 pagineIQOQ ProtocolVijay RajaindranNessuna valutazione finora
- FDA Lifecycle ApproachDocumento11 pagineFDA Lifecycle Approachsilversky09Nessuna valutazione finora
- Sample - VAL004 Req For Val Plans Protocols 7 Mar 06Documento3 pagineSample - VAL004 Req For Val Plans Protocols 7 Mar 06nagarajs50Nessuna valutazione finora
- 2015 Validation Protocol TemplateDocumento8 pagine2015 Validation Protocol TemplateJitesh M Nair100% (1)
- EU GMP Requirements-Quality SystemsDocumento70 pagineEU GMP Requirements-Quality Systemsishtidu34100% (1)
- CIQA Installation and Operational Qualification Protocol IOQ Equipment TemplateDocumento10 pagineCIQA Installation and Operational Qualification Protocol IOQ Equipment TemplateChirag prajapatiNessuna valutazione finora
- 4 - Case Study On A Risk-Based Approach To Validation - For ReviewDocumento49 pagine4 - Case Study On A Risk-Based Approach To Validation - For Reviewpate malabanan100% (1)
- Q Pharma Quality ManualDocumento32 pagineQ Pharma Quality Manualsappz354544883% (6)
- Ema Process ValidationDocumento15 pagineEma Process Validationdrs_mdu48100% (1)
- Validation Summary Report Template PDFDocumento12 pagineValidation Summary Report Template PDFAnonymous OreX6RrRP50% (2)
- Process Validation Report - RajanDocumento16 pagineProcess Validation Report - RajanRAJAN RAMANUJ67% (3)
- VMP Guide PDFDocumento6 pagineVMP Guide PDFsitimunawarohNessuna valutazione finora
- Basics of Equipment Qualification - Pharma Pathway PDFDocumento6 pagineBasics of Equipment Qualification - Pharma Pathway PDFJ VENKATESHNessuna valutazione finora
- Day 1 Session 2 - QRM Applied To CQVDocumento42 pagineDay 1 Session 2 - QRM Applied To CQVBerna Saefullah100% (1)
- GMP Supplier Assessment QuestionnaireDocumento2 pagineGMP Supplier Assessment Questionnairedrs_mdu48Nessuna valutazione finora
- Computer System Validation in The Perspective of T PDFDocumento7 pagineComputer System Validation in The Perspective of T PDFFkNessuna valutazione finora
- Validation Plan TemplateDocumento23 pagineValidation Plan Templatedes50% (2)
- Computer System Validation Definition and RequirementsDocumento3 pagineComputer System Validation Definition and RequirementsmeongNessuna valutazione finora
- Qualification of Equipment - A Risk-Based ApproachDocumento6 pagineQualification of Equipment - A Risk-Based ApproachJorge Humberto Herrera100% (9)
- Sop QualificationDocumento9 pagineSop Qualificationjohn100% (1)
- Master Validation PlanDocumento23 pagineMaster Validation Planchennamareddysriniva89% (9)
- Validation of HPLC Techniques For Pharmaceutical AnalysisDocumento17 pagineValidation of HPLC Techniques For Pharmaceutical AnalysisAhmad Abdullah Najjar100% (14)
- Guideline On General Principles of Process ValidationDocumento15 pagineGuideline On General Principles of Process ValidationRambabu komati - QANessuna valutazione finora
- Guideline For The Transfer of Analytical Test ProceduresDocumento5 pagineGuideline For The Transfer of Analytical Test Proceduresjljimenez1969100% (5)
- HR Policies 180Documento10 pagineHR Policies 180nomi1975100% (2)
- 10 Minutes With DR Abdul J KalamDocumento11 pagine10 Minutes With DR Abdul J KalamArun Kumar KanaujiaNessuna valutazione finora
- Site Master FileDocumento6 pagineSite Master FileRambabu komati - QA100% (3)
- The Basis Facts of Cleaning ValidationDocumento8 pagineThe Basis Facts of Cleaning Validationjljimenez1969100% (1)
- Guideline On Dossier Requirements For Type - 1A N 1BDocumento40 pagineGuideline On Dossier Requirements For Type - 1A N 1BRambabu komati - QA100% (1)
- A WHO GuideDocumento100 pagineA WHO Guidej.k.kumar92% (13)
- Nees Emea GuidanceDocumento31 pagineNees Emea GuidancenkszoneNessuna valutazione finora
- Esubmissions Requirements New Applications 1Documento2 pagineEsubmissions Requirements New Applications 1Rambabu komati - QANessuna valutazione finora
- Quality eCTD SubmissionsDocumento7 pagineQuality eCTD SubmissionsRambabu komati - QA100% (6)
- HR BestPracticesDocumento62 pagineHR BestPracticessaravanans100% (10)
- Froms Guide Lines 150Documento5 pagineFroms Guide Lines 150Rambabu komati - QANessuna valutazione finora
- Method ValidationDocumento16 pagineMethod ValidationRambabu komati - QA100% (3)
- Analytical Procedure Validation Manual 041 SampleDocumento3 pagineAnalytical Procedure Validation Manual 041 SampleRambabu komati - QANessuna valutazione finora
- Analytical Method ValidationDocumento86 pagineAnalytical Method ValidationRambabu komati - QA100% (12)
- Top 10 Deficiencies of Dossiers - EDQMDocumento4 pagineTop 10 Deficiencies of Dossiers - EDQMRambabu komati - QA100% (4)
- The Ten Principles of GMPDocumento3 pagineThe Ten Principles of GMPRambabu komati - QA100% (3)
- HPLC ValidationDocumento15 pagineHPLC ValidationRambabu komati - QA100% (5)
- TQMDocumento4 pagineTQMRambabu komati - QANessuna valutazione finora
- Col CareDocumento5 pagineCol Care829255Nessuna valutazione finora
- Quality Management HandbookDocumento8 pagineQuality Management HandbookRambabu komati - QA100% (2)
- 2007 QualityDocumento4 pagine2007 QualityRambabu komati - QANessuna valutazione finora
- Requirement For Service QualityDocumento14 pagineRequirement For Service QualityRambabu komati - QANessuna valutazione finora
- Quality Management HandbookDocumento8 pagineQuality Management HandbookRambabu komati - QA100% (2)
- Ten Commandments of GMPDocumento1 paginaTen Commandments of GMPRambabu komati - QA100% (3)
- Shortcuts To SuccessDocumento1 paginaShortcuts To SuccessRambabu komati - QANessuna valutazione finora
- Quality ManualDocumento32 pagineQuality ManualRambabu komati - QANessuna valutazione finora
- 2007 QualityDocumento4 pagine2007 QualityRambabu komati - QANessuna valutazione finora
- Clinical Data Management, IInd EditionDocumento18 pagineClinical Data Management, IInd EditionThomas Eipe100% (1)
- Airworthiness Inspector ManualDocumento37 pagineAirworthiness Inspector ManualDennis Padec Bwochengo100% (2)
- 5 GMP Case Studies Including FDA Analysis PDFDocumento10 pagine5 GMP Case Studies Including FDA Analysis PDFAbdul KalimNessuna valutazione finora
- Supplier - EM - HACCP - Manual - JULY 2019 PDFDocumento95 pagineSupplier - EM - HACCP - Manual - JULY 2019 PDFAkhil Singh Kushwaha100% (1)
- Microbiology Best Laboratory PracticesDocumento47 pagineMicrobiology Best Laboratory PracticesQAV_CRS100% (1)
- Checklist of Self Inspection (Quality Compliance) : Biotechnology Derived Product Facility (Unit-1)Documento4 pagineChecklist of Self Inspection (Quality Compliance) : Biotechnology Derived Product Facility (Unit-1)DebasishNessuna valutazione finora
- Presented By: Samiksha B.Sawant M, PHARM (IP), 1 SEM Guided By: Dr. Indira ParabDocumento46 paginePresented By: Samiksha B.Sawant M, PHARM (IP), 1 SEM Guided By: Dr. Indira ParabKainat MalikNessuna valutazione finora
- Brief Covering Letter - MR - Ramesh Babu - 04 07 2021Documento1 paginaBrief Covering Letter - MR - Ramesh Babu - 04 07 2021bhoopathiNessuna valutazione finora
- An Overview On Cleaning ValidationDocumento5 pagineAn Overview On Cleaning Validationedgar palominoNessuna valutazione finora
- Introducing Oct 14 PseDocumento12 pagineIntroducing Oct 14 PsekbhattacNessuna valutazione finora
- Procedure On Handling of Complaints and AppealsDocumento10 pagineProcedure On Handling of Complaints and Appealsndayiragije JMVNessuna valutazione finora
- Health Insurance System - SRDDocumento28 pagineHealth Insurance System - SRDayoobNessuna valutazione finora
- P103 - Policy On Estimating Measurement Uncertainty For Testing Laboratories-5647-3Documento4 pagineP103 - Policy On Estimating Measurement Uncertainty For Testing Laboratories-5647-3Alberto GarciaNessuna valutazione finora
- GMPSADocumento6 pagineGMPSAJames TingNessuna valutazione finora
- Safety Circuit DesignDocumento63 pagineSafety Circuit DesignMansoor KhaderNessuna valutazione finora
- Module 8 Empowered To Take Off FinalDocumento14 pagineModule 8 Empowered To Take Off FinalBilly Joe DG Dajac100% (1)
- Xiltrix BrochureDocumento8 pagineXiltrix BrochureAheesha J VadaviNessuna valutazione finora
- DCMAISO9001 AS9100 ChecklistDocumento73 pagineDCMAISO9001 AS9100 ChecklistNestor Czerwacki0% (3)
- Audit Check List (ISO 9001)Documento12 pagineAudit Check List (ISO 9001)morshed_mahamud7055Nessuna valutazione finora
- Application of EN50126 in Development of KavachDocumento16 pagineApplication of EN50126 in Development of KavachPAVAN KUMAR GUDAVALLETINessuna valutazione finora
- Monitoring and Measurement of QmsDocumento3 pagineMonitoring and Measurement of QmsamoNessuna valutazione finora
- Sis PDFDocumento2 pagineSis PDFsteam100deg1658Nessuna valutazione finora
- ISO 9001 Internal Audit TSRDocumento51 pagineISO 9001 Internal Audit TSRChan Chee LeongNessuna valutazione finora
- ISO 13485 2016 Introduction SessionDocumento100 pagineISO 13485 2016 Introduction SessionFuadi Farhana100% (1)
- IQA Checklist - SmpleDocumento16 pagineIQA Checklist - SmpleHarits As Siddiq100% (1)
- Medical Device Process Validation Plans - Oriel STAT A MATRIXDocumento5 pagineMedical Device Process Validation Plans - Oriel STAT A MATRIXGonzalo MazaNessuna valutazione finora
- Climate ChangeDocumento13 pagineClimate ChangeMoh_ElberryNessuna valutazione finora
- Implementation Haccp Food Safety System: Create and Prepare by Irwan Arief Irwan - Arief87@yahoo - Co.idDocumento40 pagineImplementation Haccp Food Safety System: Create and Prepare by Irwan Arief Irwan - Arief87@yahoo - Co.idRico Fernando TNessuna valutazione finora
- Tripura State Electricity Corporation LTDDocumento7 pagineTripura State Electricity Corporation LTDAnonymous nF09cqwNKNessuna valutazione finora
- ScaffoldingDocumento2 pagineScaffoldingBSFNessuna valutazione finora