Potrebbero piacerti anche
- Equalizer Design ExampleDocumento10 pagineEqualizer Design ExamplehaimanhptitNessuna valutazione finora
- EjercicosDocumento8 pagineEjercicosdavidNessuna valutazione finora
- 130nm BulkDocumento3 pagine130nm Bulkunseen worldNessuna valutazione finora
- Ece 346 Lab 4Documento6 pagineEce 346 Lab 4LordLawnmowerNessuna valutazione finora
- Tabla de Datos Experimentales Experimento Tiempo (Min) Vol. HCL (ML) 1 2 3 4 5 6 7 8 9 10Documento4 pagineTabla de Datos Experimentales Experimento Tiempo (Min) Vol. HCL (ML) 1 2 3 4 5 6 7 8 9 10DenisRogerNessuna valutazione finora
- DAD1 A, Sig 254,4 Ref Off (CHEM112 - 211 - 7S - A7.D)Documento100 pagineDAD1 A, Sig 254,4 Ref Off (CHEM112 - 211 - 7S - A7.D)Ucb BioeNessuna valutazione finora
- CH 3 Part 2 Tutorial - May20Documento15 pagineCH 3 Part 2 Tutorial - May20Scorpion RoyalNessuna valutazione finora
- HW2 Ans RevDocumento14 pagineHW2 Ans RevanonymoussionNessuna valutazione finora
- Superintend by D.foad: Ministry of Higher Education and Scientific ResearchDocumento7 pagineSuperintend by D.foad: Ministry of Higher Education and Scientific Researchali najatNessuna valutazione finora
- Fender Base Plate Analysis: Static CaseDocumento7 pagineFender Base Plate Analysis: Static CaseBhavesh ShiyaniNessuna valutazione finora
- Equation Solving ExamplesDocumento10 pagineEquation Solving Examplesalif08042012Nessuna valutazione finora
- Equation Solving ExamplesDocumento10 pagineEquation Solving Examplesalif08042012Nessuna valutazione finora
- GMM FinalDocumento7 pagineGMM FinalIntan Masyitha IrwanNessuna valutazione finora
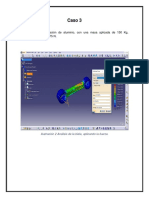
- Caso 3Documento9 pagineCaso 3ramiroNessuna valutazione finora
- An Example of The Backward Elimination ProcessDocumento8 pagineAn Example of The Backward Elimination ProcessMohammedseid AhmedinNessuna valutazione finora
- Econometrics Hina NazDocumento17 pagineEconometrics Hina NazHina Naz (F-Name :Khadim Hussain)Nessuna valutazione finora
- Zub Indeterminate Truss DegDocumento13 pagineZub Indeterminate Truss DegAnonymous jmo0bcjAsNessuna valutazione finora
- Solutions Manual To Accompany A Second Course in Statistics Regression Analysis 7th Edition 0321691695Documento47 pagineSolutions Manual To Accompany A Second Course in Statistics Regression Analysis 7th Edition 0321691695RhondaCookqjdyg100% (84)
- EE115 Solution To Prob 3 and 5 Per Unit AnalysisDocumento12 pagineEE115 Solution To Prob 3 and 5 Per Unit AnalysisJoshua Roberto GrutaNessuna valutazione finora
- Negara Maju UpdDocumento10 pagineNegara Maju UpdKapibaraNessuna valutazione finora
- MuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelDocumento8 pagineMuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelMohammad Zaky AzhariNessuna valutazione finora
- 180 NM PTM BSIM3Documento3 pagine180 NM PTM BSIM3Kshitij Goel0% (1)
- Lab 1Documento7 pagineLab 1Kashif hussainNessuna valutazione finora
- FeareportDocumento7 pagineFeareportapi-404637171Nessuna valutazione finora
- Long Report Exp 6Documento6 pagineLong Report Exp 6Mxokzah Cmoh100% (1)
- Linear Programming - Simplex Vs Interior-2-10Documento9 pagineLinear Programming - Simplex Vs Interior-2-10Fan ClubNessuna valutazione finora
- AS Practice Exercise 3 Model Solution PDFDocumento2 pagineAS Practice Exercise 3 Model Solution PDFRohan KanungoNessuna valutazione finora
- PB Mod 2 1 Homework 1 LL 2016 HRDocumento8 paginePB Mod 2 1 Homework 1 LL 2016 HRAntonio ChissanoNessuna valutazione finora
- Aa + BB +CC+DD RR A+ (B/a) B + (C/a) C + (D/a) D (R/a) R R - KC C C C N N (1-X) N N - (B/a) (N - N) N N - (C/a) (N - N) N N - (D/a) (N - N) C N /VDocumento7 pagineAa + BB +CC+DD RR A+ (B/a) B + (C/a) C + (D/a) D (R/a) R R - KC C C C N N (1-X) N N - (B/a) (N - N) N N - (C/a) (N - N) N N - (D/a) (N - N) C N /VChemical EngineeringNessuna valutazione finora
- Tugas PotensiometriDocumento4 pagineTugas PotensiometriNandia Salsa Restu MulyaniNessuna valutazione finora
- Ministry of Higher Education and Scientific Research: Kirkuk Civil Engineering Stage - ThreeDocumento8 pagineMinistry of Higher Education and Scientific Research: Kirkuk Civil Engineering Stage - Threeali najatNessuna valutazione finora
- Metode Linear BergandaDocumento16 pagineMetode Linear BergandaEka Fatihatul HikmahNessuna valutazione finora
- OTK KristiantyDocumento9 pagineOTK KristiantyDewi RatnasariNessuna valutazione finora
- Nmos180 LibDocumento2 pagineNmos180 LibKaramveer SinghNessuna valutazione finora
- EXAMPLE 8.32: N-HeptaneDocumento7 pagineEXAMPLE 8.32: N-HeptaneAria DarmawanNessuna valutazione finora
- Id Sepallengthcm Sepalwidthcm Petallengthcm Petalwidthcm Species 0 1 2 3 4Documento4 pagineId Sepallengthcm Sepalwidthcm Petallengthcm Petalwidthcm Species 0 1 2 3 4Sebastian CallesNessuna valutazione finora
- Ejercicio Numero 4Documento8 pagineEjercicio Numero 4natanael quiñoneNessuna valutazione finora
- Assignment 2.solutionDocumento13 pagineAssignment 2.solutionteweldeNessuna valutazione finora
- Machine Design ProblemsDocumento10 pagineMachine Design ProblemsSiddharth ShekharNessuna valutazione finora
- Rotación de PavimentoDocumento7 pagineRotación de PavimentoIván Pimentel OrtegaNessuna valutazione finora
- Caso 1Documento9 pagineCaso 1ramiroNessuna valutazione finora
- Impedance and Reactance: Faculty of Science Department of Physics Physics Lab 112Documento4 pagineImpedance and Reactance: Faculty of Science Department of Physics Physics Lab 112issa alatrashNessuna valutazione finora
- TekresDocumento3 pagineTekresMohammad Rayhan Kim Abdul SalamNessuna valutazione finora
- T2 3 PDFDocumento3 pagineT2 3 PDFTravis BickleNessuna valutazione finora
- UntitledDocumento8 pagineUntitledLucas Hernández Karla BereniceNessuna valutazione finora
- Component y 1. Apparent Molecular Weight (Ma) Mi Yi C1 C2 C3 n-C4 n-C5 C6 P C7+ T Jumlah RDocumento26 pagineComponent y 1. Apparent Molecular Weight (Ma) Mi Yi C1 C2 C3 n-C4 n-C5 C6 P C7+ T Jumlah RClaviano LeiwakabessyNessuna valutazione finora
- QTBDocumento7 pagineQTBUmarNessuna valutazione finora
- Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions ManualDocumento25 pagineApplied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions ManualJosephCraiggmax100% (52)
- Dwnload Full Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions Manual PDFDocumento36 pagineDwnload Full Applied Numerical Methods Wmatlab For Engineers and Scientists 3rd Edition Chapra Solutions Manual PDFeyepieceinexact.4h0h100% (11)
- Tristan Camilleri - SOR0511 - Questions - 2021.06.20Documento33 pagineTristan Camilleri - SOR0511 - Questions - 2021.06.20TristanCamilleriNessuna valutazione finora
- Natural Gas Homework2Documento42 pagineNatural Gas Homework2Khanz KhanNessuna valutazione finora
- יניבDocumento9 pagineיניבramikNessuna valutazione finora
- Final Group Report Exp 2 A4Documento5 pagineFinal Group Report Exp 2 A4nonononowayNessuna valutazione finora
- Structural Design of Two Storey Recidencnce Owner: Ferdinand Dela Cruz Address: Sto. Tomas, San Jose CityDocumento11 pagineStructural Design of Two Storey Recidencnce Owner: Ferdinand Dela Cruz Address: Sto. Tomas, San Jose CityVic ValdezNessuna valutazione finora
- Superintend by D.foad: Ministry of Higher Education and Scientific ResearchDocumento7 pagineSuperintend by D.foad: Ministry of Higher Education and Scientific ResearchAhmed falahNessuna valutazione finora
- The Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionDa EverandThe Spectra and Dynamics of Diatomic Molecules: Revised and Enlarged EditionNessuna valutazione finora
- Distributed Model Predictive Control for Plant-Wide SystemsDa EverandDistributed Model Predictive Control for Plant-Wide SystemsNessuna valutazione finora
- Modern Borehole Analytics: Annular Flow, Hole Cleaning, and Pressure ControlDa EverandModern Borehole Analytics: Annular Flow, Hole Cleaning, and Pressure ControlNessuna valutazione finora
- Networks and Graphs: Techniques and Computational MethodsDa EverandNetworks and Graphs: Techniques and Computational MethodsNessuna valutazione finora
- Sio 2Documento6 pagineSio 2Missiles dodgedNessuna valutazione finora
- Materials Letters: Ovidiu Chiscan, Ioan Dumitru, Petronel Postolache, Vasile Tura, Alexandru StancuDocumento4 pagineMaterials Letters: Ovidiu Chiscan, Ioan Dumitru, Petronel Postolache, Vasile Tura, Alexandru StancuMissiles dodgedNessuna valutazione finora
- Microwave Absorptions of Ultrathin Conductive Films and Designs of Frequency-Independent Ultrathin AbsorbersDocumento7 pagineMicrowave Absorptions of Ultrathin Conductive Films and Designs of Frequency-Independent Ultrathin AbsorbersMissiles dodgedNessuna valutazione finora
- Greenhouse Gas Emissions From A Typical Passenger VehicleDocumento5 pagineGreenhouse Gas Emissions From A Typical Passenger VehicleMissiles dodgedNessuna valutazione finora
- Nuclear Research ArticleDocumento8 pagineNuclear Research ArticleMissiles dodgedNessuna valutazione finora
- Atomic Packing Factor - Wikipedia, The Free EncyclopediaDocumento2 pagineAtomic Packing Factor - Wikipedia, The Free Encyclopediadonodoni0008Nessuna valutazione finora
- Ultrahigh Strain and Piezoelectric Behavior in Relaxor Based Ferroelectric Single CrystalsDocumento9 pagineUltrahigh Strain and Piezoelectric Behavior in Relaxor Based Ferroelectric Single CrystalsgeansoNessuna valutazione finora
- Chap 10Documento38 pagineChap 10Yusra QureshiNessuna valutazione finora
- 1993 - Avshalom C. Elitzur, Lev Vaidman - Quantum Mechanical Interaction-Free Measurements BOMBADocumento14 pagine1993 - Avshalom C. Elitzur, Lev Vaidman - Quantum Mechanical Interaction-Free Measurements BOMBAGxilleNessuna valutazione finora
- Lecture 18 - Phase Transformations - July 7 2022Documento18 pagineLecture 18 - Phase Transformations - July 7 2022Ryan MaxwellNessuna valutazione finora
- EE311A 2021 AV Slides L20Documento12 pagineEE311A 2021 AV Slides L20Ananya AgarwalNessuna valutazione finora
- PSSB 201800427Documento7 paginePSSB 201800427D SNessuna valutazione finora
- THZ WaveguidesDocumento26 pagineTHZ WaveguidesKammara BharathkumarNessuna valutazione finora
- Acta MaterialiaDocumento14 pagineActa MaterialiaSapandeepNessuna valutazione finora
- Liquid Crystalline PolymersDocumento19 pagineLiquid Crystalline PolymersKeng Goy PlungpongpanNessuna valutazione finora
- Valence Electrons For ElementsDocumento31 pagineValence Electrons For ElementsVinluan, Kaye Andrei P.Nessuna valutazione finora
- Transistors Ucp ADocumento16 pagineTransistors Ucp Azaryabimran222Nessuna valutazione finora
- Effective Values of Critical Resolved Shear Stress For Slip in Polycrystalline Magnesium and Other HCP MetalsDocumento4 pagineEffective Values of Critical Resolved Shear Stress For Slip in Polycrystalline Magnesium and Other HCP MetalsSourav MishraNessuna valutazione finora
- Alberti PhdthesisDocumento234 pagineAlberti PhdthesisUsman AliNessuna valutazione finora
- Bhushan 2009Documento9 pagineBhushan 2009Fatima BanoNessuna valutazione finora
- Chemical Bonding and Molecular Structure - JEE Main 2023 April Chapterwise PYQ - MathonGoDocumento11 pagineChemical Bonding and Molecular Structure - JEE Main 2023 April Chapterwise PYQ - MathonGokp28120724Nessuna valutazione finora
- Solidification AND Microstructure of Cast Dental AlloysDocumento97 pagineSolidification AND Microstructure of Cast Dental AlloysJASPREETKAUR0410Nessuna valutazione finora
- Ele 146Documento161 pagineEle 146Muhaned MustafaNessuna valutazione finora
- Structural Probes For Solid Materials: Dr. Graeme Blake Room 17:0015 Email G.r.blake@rug - NLDocumento97 pagineStructural Probes For Solid Materials: Dr. Graeme Blake Room 17:0015 Email G.r.blake@rug - NLAzharNessuna valutazione finora
- Solar CellDocumento3 pagineSolar CellET163018 Khaled ArjuNessuna valutazione finora
- PH3202 Physics For Electrical EngineeringDocumento5 paginePH3202 Physics For Electrical EngineeringNaWin NKNessuna valutazione finora
- Physics 73 3rd Long Exam Practice Exam 1 - The Wave Nature of Particles Practice TestDocumento7 paginePhysics 73 3rd Long Exam Practice Exam 1 - The Wave Nature of Particles Practice TestKelvin LabarezNessuna valutazione finora
- Properties of LiquidsDocumento32 pagineProperties of LiquidsSilhouette DreamNessuna valutazione finora
- Ladder Operators and The Quantum Harmonic OscillatorDocumento4 pagineLadder Operators and The Quantum Harmonic OscillatorWilliam TalmadgeNessuna valutazione finora
- Course Handout F214Documento2 pagineCourse Handout F214Aakarsh ShuklaNessuna valutazione finora
- Suspensions Colloids and Solutions WorksheetDocumento2 pagineSuspensions Colloids and Solutions WorksheetChok MallareNessuna valutazione finora
- MSE101 Final Examination 2018 AnswerDocumento14 pagineMSE101 Final Examination 2018 Answerعبدالملك جمالNessuna valutazione finora
- Vacuum Energy An Electric SystemsDocumento3 pagineVacuum Energy An Electric SystemsMarcus ReidNessuna valutazione finora
- E N.H.FDocumento2 pagineE N.H.FNaraNessuna valutazione finora