Potrebbero piacerti anche
- Ciclo RankineDocumento36 pagineCiclo RankineSara GioacchiniNessuna valutazione finora
- Gas Liquido ToDocumento7 pagineGas Liquido Todaicazzo1Nessuna valutazione finora
- 9 - Progetto Del PiattoDocumento32 pagine9 - Progetto Del PiattoAnna NunziataNessuna valutazione finora
- 4 - Concentratori o Evaporatori (Scambio Termico 2)Documento22 pagine4 - Concentratori o Evaporatori (Scambio Termico 2)teresaNessuna valutazione finora
- 8 - AssorbimentoDocumento9 pagine8 - AssorbimentoteresaNessuna valutazione finora
- EvaporazioneDocumento28 pagineEvaporazionePatricia LunguNessuna valutazione finora
- 10 - Scambiatori Di Calore (Scambio Termico 1)Documento47 pagine10 - Scambiatori Di Calore (Scambio Termico 1)Luigi SantorelliNessuna valutazione finora
- Relazione CompletaDocumento39 pagineRelazione CompletaManfre Caruso100% (3)
- La GassificazioneDocumento26 pagineLa GassificazioneLuca Fattorelli100% (1)
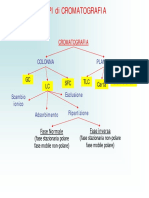
- HPLC PDFDocumento45 pagineHPLC PDFAlessandro Axl BellapiantaNessuna valutazione finora
- Static ADocumento2 pagineStatic ASilvia PalanoNessuna valutazione finora
- Analisi Volumetrica Acido - Base BiolDocumento40 pagineAnalisi Volumetrica Acido - Base BiolGio FioNessuna valutazione finora
- Ciclo FrigoriferoDocumento3 pagineCiclo FrigoriferoGiuliano Musumeci100% (1)
- Assorbimento e StrippingDocumento61 pagineAssorbimento e StrippingalessandroNessuna valutazione finora
- Concentrazione Micellare Critica 1Documento4 pagineConcentrazione Micellare Critica 1Andrea CrocettaNessuna valutazione finora
- Processo Di Denitrificazione in Impianto Di Trattamento Delle Acque ReflueDocumento47 pagineProcesso Di Denitrificazione in Impianto Di Trattamento Delle Acque RefluebeppeNessuna valutazione finora
- Assorbimento e Stripping 21.3.18Documento21 pagineAssorbimento e Stripping 21.3.18jeinerNessuna valutazione finora
- 50 Esempi Di PROBLEMIDocumento21 pagine50 Esempi Di PROBLEMIGiovanni Di MeglioNessuna valutazione finora
- Tesi Parolini Taglialatela (21-07-2010) PDFDocumento181 pagineTesi Parolini Taglialatela (21-07-2010) PDFRobertoForceNessuna valutazione finora
- AdsorbimentoDocumento10 pagineAdsorbimentoValerio CurcioNessuna valutazione finora
- Purificazione Del Gas Di SintesiDocumento14 paginePurificazione Del Gas Di SintesiMarioNessuna valutazione finora
- Università Degli Studi Di Firenze - L'aerazione Negli Impianti Di Trattamento Delle Acque ReflueDocumento67 pagineUniversità Degli Studi Di Firenze - L'aerazione Negli Impianti Di Trattamento Delle Acque ReflueunconformistNessuna valutazione finora
- Sanitaria 05 04 2019Documento21 pagineSanitaria 05 04 2019Calogero MistrettaNessuna valutazione finora
- Parte 17 DistDocumento19 pagineParte 17 DistRuslàn S.Nessuna valutazione finora
- 5-Operazioni FluidiDocumento26 pagine5-Operazioni Fluidiingfranz79Nessuna valutazione finora
- Purificazione Del Gas Di SintesiDocumento5 paginePurificazione Del Gas Di SintesiRossellaAnnelioNessuna valutazione finora
- Lezione 4 Ing. SanitariaDocumento11 pagineLezione 4 Ing. SanitariaLalaNessuna valutazione finora
- CampioneDocumento11 pagineCampioneDeejayLupinNessuna valutazione finora
- Corso HPLC Prima ParteDocumento16 pagineCorso HPLC Prima ParteAntonio Barbierotto100% (1)
- Sanitaria 08 04 2019Documento31 pagineSanitaria 08 04 2019Calogero MistrettaNessuna valutazione finora
- 3 Evaporatori-ConcentratoriDocumento14 pagine3 Evaporatori-ConcentratoriEdLardo GaleanoNessuna valutazione finora
- L'assorbimento e Lo StrippaggioDocumento3 pagineL'assorbimento e Lo Strippaggiogrecoanastasia731Nessuna valutazione finora
- Generatori Di VaporeDocumento29 pagineGeneratori Di VaporeRiccardo AgnorelliNessuna valutazione finora
- AssorbimentoDocumento42 pagineAssorbimentoSandro GoisisNessuna valutazione finora
- Generatori A VaporeDocumento30 pagineGeneratori A Vaporematealoredana8015Nessuna valutazione finora
- ImpiantiDocumento87 pagineImpiantiSimone MansiNessuna valutazione finora
- Processo Di Assorbimento (14marzo)Documento6 pagineProcesso Di Assorbimento (14marzo)Valerio CurcioNessuna valutazione finora
- Trasferimento Di MateriaDocumento56 pagineTrasferimento Di Materianp484Nessuna valutazione finora
- pr200 IntroduzioneDocumento1 paginapr200 IntroduzioneValentina FocosoNessuna valutazione finora
- Perdite Di CaricoDocumento22 paginePerdite Di CaricoantoniodellisantiNessuna valutazione finora
- Em 05Documento25 pagineEm 05francoNessuna valutazione finora
- Flottazione PDFDocumento7 pagineFlottazione PDFNick3105870000Nessuna valutazione finora
- 5 - DistillazioneDocumento64 pagine5 - DistillazioneRaffaella Nunziata100% (1)
- Impianti A Fanghi AttiviDocumento21 pagineImpianti A Fanghi AttiviLuca PataunerNessuna valutazione finora
- Università Degli Studi Di Udine - La Respirometria Del Fango Attivo PDFDocumento52 pagineUniversità Degli Studi Di Udine - La Respirometria Del Fango Attivo PDFunconformistNessuna valutazione finora
- 3 - Polimerizzazione Dello Stirene - RadicalicaDocumento12 pagine3 - Polimerizzazione Dello Stirene - RadicalicaLuca Marletta100% (2)
- Impianti Meccanici CompletoDocumento78 pagineImpianti Meccanici CompletoMichele AssirelliNessuna valutazione finora
- Modellistica Dei Combustori - (D. Lentini)Documento110 pagineModellistica Dei Combustori - (D. Lentini)pezz07Nessuna valutazione finora
- Cicli CombinatiDocumento30 pagineCicli CombinatiNiccolò Giannotti100% (3)
- CicloniDocumento28 pagineCiclonicastibra100% (2)
- Metodo PSDocumento6 pagineMetodo PSValerio CurcioNessuna valutazione finora
- Acido AceticoDocumento35 pagineAcido AceticoAmalia RamunnoNessuna valutazione finora
- Foronomia 11ottobreDocumento12 pagineForonomia 11ottobreDavid SmithNessuna valutazione finora
- Scambiatori Di CaloreDocumento24 pagineScambiatori Di CaloreFILIPPONessuna valutazione finora
- HPLC (Analisi Farmaceutica)Documento4 pagineHPLC (Analisi Farmaceutica)eleonoramaria.vacchini01Nessuna valutazione finora
- Rene 3Documento20 pagineRene 3api-3704348100% (1)
- ORC BidiniDocumento6 pagineORC BidinilunettinoNessuna valutazione finora
- Riassunto PDFDocumento484 pagineRiassunto PDFgiuNessuna valutazione finora
- Fluidodinamica Delle Macchine Introduzione Agli Efflussi Nelle Turbomacchine e Alla Teoria Della SimilitudineDocumento127 pagineFluidodinamica Delle Macchine Introduzione Agli Efflussi Nelle Turbomacchine e Alla Teoria Della SimilitudinetundolamiNessuna valutazione finora
- Petronio (2011) PHD T - Numerical Investigation of Condensation and Evaporation Effects Inside A TubDocumento126 paginePetronio (2011) PHD T - Numerical Investigation of Condensation and Evaporation Effects Inside A Tubfs.cNessuna valutazione finora
- Catalizzatori Di Ossidazione 20-11Documento5 pagineCatalizzatori Di Ossidazione 20-11Valerio CurcioNessuna valutazione finora
- Diagrammi Di Stato - EserciziDocumento13 pagineDiagrammi Di Stato - EserciziValerio CurcioNessuna valutazione finora
- Processo Di Assorbimento (14marzo)Documento7 pagineProcesso Di Assorbimento (14marzo)Valerio CurcioNessuna valutazione finora
- Modx Manuale 2.0 PDFDocumento46 pagineModx Manuale 2.0 PDFValerio CurcioNessuna valutazione finora
- Gert-Jan Gruter Vrije Universiteit AvantiumDocumento11 pagineGert-Jan Gruter Vrije Universiteit AvantiumValerio CurcioNessuna valutazione finora
- Modx Manuale 2.0 PDFDocumento46 pagineModx Manuale 2.0 PDFValerio CurcioNessuna valutazione finora
- La Distillazione Reattiva È Una Delle Tecnologie Di Separazione ReattivaDocumento5 pagineLa Distillazione Reattiva È Una Delle Tecnologie Di Separazione ReattivaValerio CurcioNessuna valutazione finora
- Esercizi Matlab 1Documento3 pagineEsercizi Matlab 1Valerio CurcioNessuna valutazione finora
- Esercizio Pib Fermentatore BatchDocumento28 pagineEsercizio Pib Fermentatore BatchValerio CurcioNessuna valutazione finora
- AdsorbimentoDocumento12 pagineAdsorbimentoValerio CurcioNessuna valutazione finora
- Lezione Matlab 1 PDFDocumento24 pagineLezione Matlab 1 PDFValerio CurcioNessuna valutazione finora
- Metodo PSDocumento6 pagineMetodo PSValerio CurcioNessuna valutazione finora
- Esercizi Matlab 1Documento3 pagineEsercizi Matlab 1Valerio CurcioNessuna valutazione finora
- Esercizi DinamicaDocumento4 pagineEsercizi DinamicaValerio CurcioNessuna valutazione finora
- Rimozione Del Particolato 13-12Documento10 pagineRimozione Del Particolato 13-12Valerio CurcioNessuna valutazione finora
- Esercizi ControlloDocumento6 pagineEsercizi ControlloValerio CurcioNessuna valutazione finora
- DeSOx Parte 2Documento5 pagineDeSOx Parte 2Valerio CurcioNessuna valutazione finora
- Assorbimento (13-03-2018)Documento12 pagineAssorbimento (13-03-2018)Valerio CurcioNessuna valutazione finora
- Introduzione 6-11Documento9 pagineIntroduzione 6-11Valerio CurcioNessuna valutazione finora
- Introduzione 6-11Documento1 paginaIntroduzione 6-11Valerio CurcioNessuna valutazione finora
- Foglio Di Calcolo Colonne Assorbimento (HTU - NTU) (Ripristinato Automaticamente)Documento5 pagineFoglio Di Calcolo Colonne Assorbimento (HTU - NTU) (Ripristinato Automaticamente)Valerio CurcioNessuna valutazione finora
- Processi Biologici Per La Rimozione Simultanea Di Sostanza Organica e Azoto Nelle AcqueDocumento10 pagineProcessi Biologici Per La Rimozione Simultanea Di Sostanza Organica e Azoto Nelle AcqueValerio CurcioNessuna valutazione finora
- DeSOx Parte 2Documento5 pagineDeSOx Parte 2Valerio CurcioNessuna valutazione finora
- Processo Di Assorbimento (14marzo)Documento6 pagineProcesso Di Assorbimento (14marzo)Valerio CurcioNessuna valutazione finora
- 10bis. DeSOx Parte 3Documento11 pagine10bis. DeSOx Parte 3Valerio CurcioNessuna valutazione finora
- AdsorbimentoDocumento10 pagineAdsorbimentoValerio CurcioNessuna valutazione finora
- Havana Sheet MusicDocumento7 pagineHavana Sheet MusicValerio CurcioNessuna valutazione finora
- InquinamentoDocumento4 pagineInquinamentoValerio CurcioNessuna valutazione finora
- Introduzione 6-11Documento1 paginaIntroduzione 6-11Valerio CurcioNessuna valutazione finora
- Biometro Di BovisDocumento5 pagineBiometro Di BovisMondo Beat100% (1)
- Che 110 Evst PDFDocumento23 pagineChe 110 Evst PDFAlisha AgarwalNessuna valutazione finora
- Tecnica GasDocumento60 pagineTecnica Gasgennaro_basileNessuna valutazione finora
- Due Diligence - Slide RINADocumento15 pagineDue Diligence - Slide RINAMarco MacciNessuna valutazione finora
- Interpretazione Esoterica DantescaDocumento19 pagineInterpretazione Esoterica DantescadarmalaiNessuna valutazione finora
- Le NubiDocumento7 pagineLe Nubitommy_84_mail5328Nessuna valutazione finora
- Linee Guida PMA 2014Documento32 pagineLinee Guida PMA 2014Non sono MandrakeNessuna valutazione finora
- Lezione 7 Ciclo Idrologico - Proprietà SuoloDocumento63 pagineLezione 7 Ciclo Idrologico - Proprietà SuoloIvan AzzaràNessuna valutazione finora
- Sma - Off GridDocumento172 pagineSma - Off GridSilvian IonescuNessuna valutazione finora
- Zanichelli 2 0289 PreviewDocumento30 pagineZanichelli 2 0289 PreviewPeppe D. BorrelloNessuna valutazione finora
- 1 359Documento356 pagine1 359Kun Mindaugas Malinauskas SJNessuna valutazione finora