Potrebbero piacerti anche
- Clinical Approach To Neonatal JaundiceDocumento17 pagineClinical Approach To Neonatal JaundiceSabera KapasiNessuna valutazione finora
- JaundiceDocumento75 pagineJaundiceangel_sagun_1Nessuna valutazione finora
- Drug Therapy During PregnancyDa EverandDrug Therapy During PregnancyTom K. A. B. EskesValutazione: 5 su 5 stelle5/5 (1)
- Measles: Wilbert H. Mason Nelson's Textbook of Pediatrics 20th EditionDocumento30 pagineMeasles: Wilbert H. Mason Nelson's Textbook of Pediatrics 20th EditionEarl Mel Dustin LaoNessuna valutazione finora
- Nursing the NeonateDa EverandNursing the NeonateMaggie MeeksNessuna valutazione finora
- Neonatal Jaundice and Prolonged Jaundice in Newborn InfantsDocumento33 pagineNeonatal Jaundice and Prolonged Jaundice in Newborn InfantskeyRielleNessuna valutazione finora
- Contraception: Science and PracticeDa EverandContraception: Science and PracticeMarcus FilshieNessuna valutazione finora
- Obg Extra EdgeDocumento201 pagineObg Extra EdgeabhishekbmcNessuna valutazione finora
- Neonatal JaundiceDocumento18 pagineNeonatal JaundiceArinaManaSikanaNessuna valutazione finora
- Problem-based Approach to Gastroenterology and HepatologyDa EverandProblem-based Approach to Gastroenterology and HepatologyJohn N. PlevrisNessuna valutazione finora
- Neonatal JaundiceDocumento48 pagineNeonatal Jaundicelordoftheweb100% (22)
- Neonatal Jaundice CmeDocumento39 pagineNeonatal Jaundice CmeGideon K. Mutai100% (1)
- Seminar 6 Approach To Neonatal JaundiceDocumento50 pagineSeminar 6 Approach To Neonatal JaundiceKelvin Su100% (1)
- Neonatal Jaundice (Wong)Documento54 pagineNeonatal Jaundice (Wong)Siti Hajar100% (1)
- Nephroblastoma FinalDocumento24 pagineNephroblastoma FinalKrissy_Singh_211Nessuna valutazione finora
- Neonatal InfectionDocumento56 pagineNeonatal InfectionAgust SalimNessuna valutazione finora
- Heavy Menstrual BleedingDocumento29 pagineHeavy Menstrual BleedingsanjupainNessuna valutazione finora
- Neonatal JaundiceDocumento12 pagineNeonatal JaundiceAlya Afifa100% (1)
- Neonatal Jaundice DrkumarDocumento44 pagineNeonatal Jaundice Drkumarvasu_5iveNessuna valutazione finora
- Management of Neonatal JaundiceDocumento22 pagineManagement of Neonatal JaundiceSuhazeli Abdullah100% (1)
- Nephrotic SyndromeDocumento57 pagineNephrotic SyndromePradnya WartheNessuna valutazione finora
- Pathology of Female Genital Tract Short NotesDocumento5 paginePathology of Female Genital Tract Short NotesameerabestNessuna valutazione finora
- Breech Presentation: Group 3: Danuco, Demain, FernandezDocumento16 pagineBreech Presentation: Group 3: Danuco, Demain, FernandezKylene DanucoNessuna valutazione finora
- Fetal DistressDocumento2 pagineFetal DistressBal Ri Mekoleu100% (1)
- Neonatal JaundiceDocumento30 pagineNeonatal JaundiceAnonymous Y3fqBduNessuna valutazione finora
- Management of Pregnancy Related BleedingDocumento75 pagineManagement of Pregnancy Related BleedingChuah Wei Hong100% (1)
- Pyloric StenosisDocumento3 paginePyloric Stenosismagisasamundo100% (1)
- Ob10 Abnormal LabourDocumento76 pagineOb10 Abnormal LabourFazzril HasmiNessuna valutazione finora
- Exchange Blood TransfusionDocumento38 pagineExchange Blood TransfusionMeseret Hamer Zewdie100% (1)
- Neonatal Jaundice Lecture,,By DR Kassahun GirmaDocumento25 pagineNeonatal Jaundice Lecture,,By DR Kassahun GirmaKassahun Girma GelawNessuna valutazione finora
- Obs-Gynae CP Final (2nd Batch)Documento89 pagineObs-Gynae CP Final (2nd Batch)Dipesh ShresthaNessuna valutazione finora
- Infections During PregnancyDocumento9 pagineInfections During PregnancyKarina Madriaga100% (4)
- Surgery - Pediatric GIT, Abdominal Wall, Neoplasms - 2014ADocumento14 pagineSurgery - Pediatric GIT, Abdominal Wall, Neoplasms - 2014ATwinkle SalongaNessuna valutazione finora
- PUERPERIUMDocumento58 paginePUERPERIUMPencenk Azznew100% (4)
- MCQ GitDocumento15 pagineMCQ Gitpriya009100% (1)
- Albendazole in PediatricsDocumento38 pagineAlbendazole in PediatricsKishore ChandkiNessuna valutazione finora
- 6th Year Obs&Gyne 2015Documento19 pagine6th Year Obs&Gyne 2015Rashed ShatnawiNessuna valutazione finora
- Necrotizing EnterocolitisDocumento36 pagineNecrotizing EnterocolitisMahad Maxamed AxmedNessuna valutazione finora
- CASE 2 PneumoniaDocumento12 pagineCASE 2 PneumoniaKenneth MiguelNessuna valutazione finora
- Gyne LE 2: Stage IVA Is Characterized byDocumento49 pagineGyne LE 2: Stage IVA Is Characterized byĐinesh IlavarasanNessuna valutazione finora
- Approach To Neonatal JaundiceDocumento73 pagineApproach To Neonatal JaundiceG Venkatesh50% (2)
- Pneumonia in PaediatricsDocumento19 paginePneumonia in PaediatricsBhintuna ChaNessuna valutazione finora
- ContraceptionDocumento53 pagineContraceptionfiea241089100% (7)
- Hemophilia PathophysiologyDocumento79 pagineHemophilia Pathophysiologyjeba100% (8)
- Case 10: Placenta Previa: ST ND RDDocumento6 pagineCase 10: Placenta Previa: ST ND RDMengda ZhangNessuna valutazione finora
- RH Isoimmunization DR SamiraDocumento37 pagineRH Isoimmunization DR Samiragynaecology2010100% (2)
- LeukemiaDocumento33 pagineLeukemiarohan_5100% (6)
- Approach and Management of Bleeding NeonateDocumento49 pagineApproach and Management of Bleeding NeonateG VenkateshNessuna valutazione finora
- HX Taking of PeadiatricsDocumento5 pagineHX Taking of PeadiatricsCahaya Al-Hazeenillah100% (1)
- Bullous PemphigoidDocumento21 pagineBullous PemphigoidChe Ainil ZainodinNessuna valutazione finora
- Approach To Neonatal HyperbilirubinemiaDocumento34 pagineApproach To Neonatal HyperbilirubinemiaNilesh HatzadeNessuna valutazione finora
- Birth DefectsDocumento58 pagineBirth DefectsrohitNessuna valutazione finora
- An Overview of Paediatric SyndromesDocumento50 pagineAn Overview of Paediatric SyndromesSana Bushra100% (11)
- Obstetric and Clinical Medicine - Gynaecological History and ExaminationDocumento7 pagineObstetric and Clinical Medicine - Gynaecological History and ExaminationShahin Kazemzadeh100% (1)
- Nelson Pediatrics Review (MCQS) 17edDocumento631 pagineNelson Pediatrics Review (MCQS) 17edGabrielle Maycock75% (8)
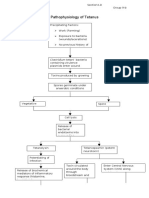
- Pathophysiology of Tetanus: Clostridium Tetani BacteriaDocumento3 paginePathophysiology of Tetanus: Clostridium Tetani BacteriaSlepy chng100% (1)
- Osta Lecture 4 Notes Online ENGLISH Base of Skull and BrainDocumento36 pagineOsta Lecture 4 Notes Online ENGLISH Base of Skull and BrainslyfoxkittyNessuna valutazione finora
- Edwards Critical Care Quick Guide To Cardiopulmonary Care PDFDocumento188 pagineEdwards Critical Care Quick Guide To Cardiopulmonary Care PDFanonNessuna valutazione finora
- Kemu A & P 2Documento6 pagineKemu A & P 2malenya1Nessuna valutazione finora
- Mechanical Loads - Bone ResponseDocumento19 pagineMechanical Loads - Bone ResponselllllAdelalllllNessuna valutazione finora
- Bio PP1 QS Pre-Mock 2024.Documento9 pagineBio PP1 QS Pre-Mock 2024.Elvis KemboiNessuna valutazione finora
- Attribution Theory and Its ApplicationsDocumento14 pagineAttribution Theory and Its Applicationsklock100% (3)
- Assessment of Fluid Responsiveness Recent Advances. OkDocumento6 pagineAssessment of Fluid Responsiveness Recent Advances. OkLeiniker Navarro Rey100% (1)
- FertilityDocumento207 pagineFertilitypmotcNessuna valutazione finora
- SanMilan Inigo Cycling Physiology and Physiological TestingDocumento67 pagineSanMilan Inigo Cycling Physiology and Physiological Testingjesus.clemente.90Nessuna valutazione finora
- Stress Management: Personal Development Grade 12Documento11 pagineStress Management: Personal Development Grade 12John Russel ColigadoNessuna valutazione finora
- HUMAN IMMUNE SYSTEM - Notes RepairedDocumento19 pagineHUMAN IMMUNE SYSTEM - Notes RepairedLoren EscotoNessuna valutazione finora
- Charge AssociationDocumento2 pagineCharge AssociationBlueash BehNessuna valutazione finora
- Interpersonal Communications Chapter 4Documento5 pagineInterpersonal Communications Chapter 4randomscribduser2012Nessuna valutazione finora
- მიტოქონდრიის ფუნქცია და კიბოDocumento1 paginaმიტოქონდრიის ფუნქცია და კიბოEMD GROUPNessuna valutazione finora
- The Kriya Yoga PracticesDocumento11 pagineThe Kriya Yoga PracticesTushar Naik100% (46)
- General Biology Q4 M7Documento19 pagineGeneral Biology Q4 M7Mubin AbdulkarilNessuna valutazione finora
- Bio1301 Lecture Note - Plant Section PDFDocumento32 pagineBio1301 Lecture Note - Plant Section PDFAjomNessuna valutazione finora
- AbscessDocumento26 pagineAbscessThusith WijayawardenaNessuna valutazione finora
- 9700 s03 QP 4Documento12 pagine9700 s03 QP 4Lâm Văn Sa HuỳnhNessuna valutazione finora
- Bristol Stool ChartDocumento5 pagineBristol Stool ChartlguerreroNessuna valutazione finora
- TramadolDocumento2 pagineTramadolPapaindoNessuna valutazione finora
- Analzying Sensory Somatic Responces - System Reinforcemnt Worksheet1Documento2 pagineAnalzying Sensory Somatic Responces - System Reinforcemnt Worksheet1Creative DestinyNessuna valutazione finora
- Arterial Blood Gas Interpretation: Joseph Brian L. Costiniano, MD, DPCPDocumento39 pagineArterial Blood Gas Interpretation: Joseph Brian L. Costiniano, MD, DPCPGio Tamaño BalisiNessuna valutazione finora
- Modified Beggs / Orthodontic Courses by Indian Dental AcademyDocumento202 pagineModified Beggs / Orthodontic Courses by Indian Dental Academyindian dental academyNessuna valutazione finora
- OB Definition of TermsDocumento9 pagineOB Definition of TermsWarrenSandovalNessuna valutazione finora
- Curricullum - Health Assessment Syllabus-1Documento4 pagineCurricullum - Health Assessment Syllabus-1KATZ2014Nessuna valutazione finora
- ReproductionDocumento48 pagineReproductionaufaaNessuna valutazione finora
- Urinary SystemDocumento31 pagineUrinary SystemAyro Business CenterNessuna valutazione finora
- IADVL Color Atlas of Dermatopathology - BookDocumento41 pagineIADVL Color Atlas of Dermatopathology - Book65gkenNessuna valutazione finora
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionDa EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionValutazione: 4 su 5 stelle4/5 (404)
- The Age of Magical Overthinking: Notes on Modern IrrationalityDa EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityValutazione: 4 su 5 stelle4/5 (34)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDDa EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDValutazione: 5 su 5 stelle5/5 (4)
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Da EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Valutazione: 3 su 5 stelle3/5 (1)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsDa EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsValutazione: 4 su 5 stelle4/5 (4)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedDa EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedValutazione: 4.5 su 5 stelle4.5/5 (82)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeDa EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeValutazione: 4.5 su 5 stelle4.5/5 (254)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsDa EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsValutazione: 4.5 su 5 stelle4.5/5 (170)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsDa EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNessuna valutazione finora
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeDa EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeValutazione: 2 su 5 stelle2/5 (1)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisDa EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisValutazione: 4.5 su 5 stelle4.5/5 (44)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsDa EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsValutazione: 5 su 5 stelle5/5 (1)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaDa EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Manipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesDa EverandManipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesValutazione: 4.5 su 5 stelle4.5/5 (1412)
- The Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeDa EverandThe Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeValutazione: 4.5 su 5 stelle4.5/5 (2)
- The Obesity Code: Unlocking the Secrets of Weight LossDa EverandThe Obesity Code: Unlocking the Secrets of Weight LossValutazione: 4 su 5 stelle4/5 (6)
- To Explain the World: The Discovery of Modern ScienceDa EverandTo Explain the World: The Discovery of Modern ScienceValutazione: 3.5 su 5 stelle3.5/5 (51)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Da EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Valutazione: 4.5 su 5 stelle4.5/5 (110)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisDa EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisValutazione: 3.5 su 5 stelle3.5/5 (2)
- The Garden Within: Where the War with Your Emotions Ends and Your Most Powerful Life BeginsDa EverandThe Garden Within: Where the War with Your Emotions Ends and Your Most Powerful Life BeginsNessuna valutazione finora
- Hearts of Darkness: Serial Killers, The Behavioral Science Unit, and My Life as a Woman in the FBIDa EverandHearts of Darkness: Serial Killers, The Behavioral Science Unit, and My Life as a Woman in the FBIValutazione: 4 su 5 stelle4/5 (20)
- Why We Die: The New Science of Aging and the Quest for ImmortalityDa EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityValutazione: 4.5 su 5 stelle4.5/5 (6)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryDa EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryValutazione: 4 su 5 stelle4/5 (46)