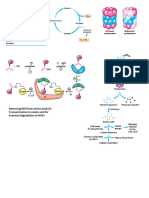
Potrebbero piacerti anche
- Bioorganic & Medicinal ChemistryDocumento9 pagineBioorganic & Medicinal ChemistryHải VânNessuna valutazione finora
- 19-Design of An Amperometric Biosensor For The Determination ofDocumento4 pagine19-Design of An Amperometric Biosensor For The Determination ofwardaninurindahNessuna valutazione finora
- Engelke1990 PDFDocumento5 pagineEngelke1990 PDFDiego GarzonNessuna valutazione finora
- Crystallization and Preliminary X-Ray Analysis of Methionyl-tRNA, FormyltransferaseDocumento3 pagineCrystallization and Preliminary X-Ray Analysis of Methionyl-tRNA, FormyltransferaseSalma OugriNessuna valutazione finora
- Proteins - Structure, Function, and Bioinformatics Volume 25 Issue 1 1996Documento3 pagineProteins - Structure, Function, and Bioinformatics Volume 25 Issue 1 1996Jhonatan Castillo SullcaNessuna valutazione finora
- Biochemistry - C4 Proteins Determination of Primary StructureDocumento2 pagineBiochemistry - C4 Proteins Determination of Primary StructureKim LlamasNessuna valutazione finora
- Ribosome-Targeting Antibiotics and Mechanisms of Bacterial ResistanceDocumento14 pagineRibosome-Targeting Antibiotics and Mechanisms of Bacterial ResistanceColeguillasNessuna valutazione finora
- Fluorescence Labeling of The C-Terminus of Proteins With A Puromycin Analogue in Cell-Free Translation SystemsDocumento4 pagineFluorescence Labeling of The C-Terminus of Proteins With A Puromycin Analogue in Cell-Free Translation SystemsFauziana NurhanisahNessuna valutazione finora
- Surface Ftir Techniques To Analyze The Conformation of Proteinspeptides in H2o Environment 2161 0398 1000202Documento8 pagineSurface Ftir Techniques To Analyze The Conformation of Proteinspeptides in H2o Environment 2161 0398 1000202S MajumderNessuna valutazione finora
- Surface-Enhanced Raman Spectroscopy Study of The Interaction of The Antitumoral Drug Emodin With Human Serum AlbuminDocumento6 pagineSurface-Enhanced Raman Spectroscopy Study of The Interaction of The Antitumoral Drug Emodin With Human Serum AlbuminMaulana Rasyid IbrahimNessuna valutazione finora
- Transcription NotesDocumento83 pagineTranscription NotesKrishna YaduvanshiNessuna valutazione finora
- Practical Synthesis of Spermine, Thermospermine and NorspermineDocumento5 paginePractical Synthesis of Spermine, Thermospermine and NorspermineFRANCIS NDOURNessuna valutazione finora
- 1 s2.0 S0141813017340424 MainDocumento8 pagine1 s2.0 S0141813017340424 MainNegreanuDenisaValentinaNessuna valutazione finora
- Div Class Title Northern Blotting DivDocumento7 pagineDiv Class Title Northern Blotting DivamarNessuna valutazione finora
- Asai Et Al 2000 Ceramide PheromonesDocumento5 pagineAsai Et Al 2000 Ceramide PheromonesharisankarhsNessuna valutazione finora
- Mar 1Documento14 pagineMar 1avmrimachemist100% (2)
- Pnas00069 0190Documento5 paginePnas00069 0190h66656440Nessuna valutazione finora
- Ricin AbrinDocumento9 pagineRicin Abrinjftfth100% (1)
- 2016 Zeitschrift Fur Anorganische Und Aligemeine ChemieDocumento6 pagine2016 Zeitschrift Fur Anorganische Und Aligemeine ChemieildaNessuna valutazione finora
- Random-Splitting of tRNA Transcripts As An Approach For Studying tRNA-protein InteractionsDocumento4 pagineRandom-Splitting of tRNA Transcripts As An Approach For Studying tRNA-protein InteractionslemNessuna valutazione finora
- Mcb150 Lecture 19 NotesDocumento5 pagineMcb150 Lecture 19 Noteskaren_duenas_6Nessuna valutazione finora
- 10 1021@jacs 9b13910Documento5 pagine10 1021@jacs 9b13910Bogdan NechitaNessuna valutazione finora
- Comparative Study For Determination of Atracurium Besilate in Presence of Its Toxic Degradant (Laudanosine) by Reversed Phase HPLC and by TLC DensitometryDocumento11 pagineComparative Study For Determination of Atracurium Besilate in Presence of Its Toxic Degradant (Laudanosine) by Reversed Phase HPLC and by TLC DensitometrySabrina JonesNessuna valutazione finora
- Analysis of Molecular FossilsDocumento4 pagineAnalysis of Molecular FossilsEssam Eldin Metwally AhmedNessuna valutazione finora
- NMR, ESI/MS, and MALDI-TOF/MS Analysis of Pear Juice Polymeric Proanthocyanidins With Potent Free Radical Scavenging ActivityDocumento9 pagineNMR, ESI/MS, and MALDI-TOF/MS Analysis of Pear Juice Polymeric Proanthocyanidins With Potent Free Radical Scavenging ActivityKimberly GutièrrezNessuna valutazione finora
- II. THE Trypanocidal Action OF Arsenic CompoundsDocumento19 pagineII. THE Trypanocidal Action OF Arsenic CompoundsAlex12Nessuna valutazione finora
- Lesson 5Documento63 pagineLesson 5charith chiranthaNessuna valutazione finora
- Echinacea Camnabinoides MimeticosDocumento5 pagineEchinacea Camnabinoides MimeticosDaryl TinjacaNessuna valutazione finora
- Two Colorimetric Ampicillin Sensing Schemes Based On The Interaction of Aptamers With Gold NanoparticlesDocumento10 pagineTwo Colorimetric Ampicillin Sensing Schemes Based On The Interaction of Aptamers With Gold Nanoparticleslox agencyNessuna valutazione finora
- Synthesis and in Vitro Antiprotozoal Activities of 5-Phenyliminobenzo (A) Phenoxazine DerivativesDocumento4 pagineSynthesis and in Vitro Antiprotozoal Activities of 5-Phenyliminobenzo (A) Phenoxazine Derivativesfaz7Nessuna valutazione finora
- Atp PDFDocumento8 pagineAtp PDFMasresha AhmedNessuna valutazione finora
- Biochemistry - Final Exam PreparationDocumento17 pagineBiochemistry - Final Exam Preparationshambhavig108Nessuna valutazione finora
- Edwards 2003Documento11 pagineEdwards 2003Arif GunawanNessuna valutazione finora
- Marini 1997Documento12 pagineMarini 1997Rodrigo AbreuNessuna valutazione finora
- Special Articles Serum Albumin: Marcus Murray Oratz Sidney S. SchreiberDocumento17 pagineSpecial Articles Serum Albumin: Marcus Murray Oratz Sidney S. SchreiberTuấn AnhNessuna valutazione finora
- 3405Documento9 pagine3405khawarkhubaibNessuna valutazione finora
- Identification of A Dipeptide UnknownDocumento13 pagineIdentification of A Dipeptide UnknownVero Herrera CaroNessuna valutazione finora
- Pyrazines From Bacteria and Ants Convergent ChemisDocumento7 paginePyrazines From Bacteria and Ants Convergent ChemisJorgeVictorMauriceLiraNessuna valutazione finora
- 12TrlAMO LicyayoDocumento9 pagine12TrlAMO LicyayoMohamidin MamalapatNessuna valutazione finora
- Researcharticle: Pemurnian Dan Karakterisasi Melanogenic Enzim Tirosinase Tombol JamurDocumento7 pagineResearcharticle: Pemurnian Dan Karakterisasi Melanogenic Enzim Tirosinase Tombol Jamuranon_853052696Nessuna valutazione finora
- FRESNO Et Al-1977-European Journal of BiochemistryDocumento8 pagineFRESNO Et Al-1977-European Journal of BiochemistryGabriel AlvesNessuna valutazione finora
- Comparison of Tryptophan Interactions To Free and Grafted BSA ProteinDocumento7 pagineComparison of Tryptophan Interactions To Free and Grafted BSA ProteinangeljosechuquiureNessuna valutazione finora
- Poly (Vinyl Methyl Ether) Poly (Methyl Methacrylate) Blends Using Diblock CopolymerDocumento5 paginePoly (Vinyl Methyl Ether) Poly (Methyl Methacrylate) Blends Using Diblock Copolymerfadhillah ivanNessuna valutazione finora
- Biosensors and Sensors For Dopamine DetectionDocumento16 pagineBiosensors and Sensors For Dopamine DetectionArjun BalaramanNessuna valutazione finora
- Mechanisms of Inhibition of Phenylalanine Ammonia-Lyase by Phenol Inhibitors and Phenol/glycine Synergistic InhibitorsDocumento6 pagineMechanisms of Inhibition of Phenylalanine Ammonia-Lyase by Phenol Inhibitors and Phenol/glycine Synergistic InhibitorsJesusNessuna valutazione finora
- 2015 - P81-Salvia UrmiensisDocumento6 pagine2015 - P81-Salvia Urmiensismehdiamohamadi67Nessuna valutazione finora
- Formaldehyde Gel Electrophoresis of Total RNADocumento4 pagineFormaldehyde Gel Electrophoresis of Total RNAGhanshyam R ParmarNessuna valutazione finora
- Mechanism "Ecstasy" (3,4-Methylenedioxy-Methamphetamine (MDMA) )Documento5 pagineMechanism "Ecstasy" (3,4-Methylenedioxy-Methamphetamine (MDMA) )nalinda avishkaNessuna valutazione finora
- Ragoussis 2007Documento3 pagineRagoussis 2007NanjundaswamyNessuna valutazione finora
- Product Name: Amphetane Phosphate Chemical PropertiesDocumento1 paginaProduct Name: Amphetane Phosphate Chemical PropertiesMarkoNessuna valutazione finora
- Rsos 172407-1 PDFDocumento13 pagineRsos 172407-1 PDFEka AgustinNessuna valutazione finora
- Jacs 1c04147 PDFDocumento5 pagineJacs 1c04147 PDFSamir SahaNessuna valutazione finora
- Draft Assigment Bio 2Documento2 pagineDraft Assigment Bio 2puteri hanisNessuna valutazione finora
- Severino 2021Documento11 pagineSeverino 2021roger souza de oliveiraNessuna valutazione finora
- RNA RNA 1 RNA RNA 40 μg / mL Mrna 2 2SEDocumento3 pagineRNA RNA 1 RNA RNA 40 μg / mL Mrna 2 2SESameerNessuna valutazione finora
- Detection of AmineDocumento6 pagineDetection of AminedumitriuNessuna valutazione finora
- Interaction Study of Amino Acid On Novel Kagome Phosphorene Nanotube - A DFT OutlookDocumento10 pagineInteraction Study of Amino Acid On Novel Kagome Phosphorene Nanotube - A DFT OutlookRanjan SutradharNessuna valutazione finora
- Spectrop Hotometric Determination of Polyquaterniu M - I With Trypan Blue by A Difference ProcedureDocumento3 pagineSpectrop Hotometric Determination of Polyquaterniu M - I With Trypan Blue by A Difference ProcedureHammam HafidzurahmanNessuna valutazione finora
- Formal Rep 2Documento4 pagineFormal Rep 2PATRICIA RAE ENDAYANessuna valutazione finora
- 2011 01 94 PDFDocumento6 pagine2011 01 94 PDFVel MuruganNessuna valutazione finora
- Olymyxins A Review Focusing On Their NephrotoxicityDocumento7 pagineOlymyxins A Review Focusing On Their NephrotoxicityVel MuruganNessuna valutazione finora
- Iaut 05 I 4 P 275Documento1 paginaIaut 05 I 4 P 275Vel MuruganNessuna valutazione finora
- Dynamic Storage Allocation in The Atlas Computer, Including An Automatic Use of A Backing StoreDocumento2 pagineDynamic Storage Allocation in The Atlas Computer, Including An Automatic Use of A Backing StoreVel MuruganNessuna valutazione finora
- Notice To ShareholdersDocumento6 pagineNotice To ShareholdersVel MuruganNessuna valutazione finora
- ICI Application Well Built Residential AwardsDocumento6 pagineICI Application Well Built Residential AwardsVel MuruganNessuna valutazione finora
- Steel Placement HandbookDocumento56 pagineSteel Placement HandbookVel MuruganNessuna valutazione finora
- Hakushima Hiroshima Sta. Yokogawa StaDocumento1 paginaHakushima Hiroshima Sta. Yokogawa StaVel MuruganNessuna valutazione finora
- Physicochemical Analysis of Triveni Lake Water of Amravati District in (MS) IndiaDocumento3 paginePhysicochemical Analysis of Triveni Lake Water of Amravati District in (MS) IndiaVel MuruganNessuna valutazione finora
- 16 1 Stone PDFDocumento29 pagine16 1 Stone PDFVel MuruganNessuna valutazione finora
- Ej 047 Pages 025 034Documento10 pagineEj 047 Pages 025 034Vel MuruganNessuna valutazione finora
- Reinforcing Steel Placement Handbook: NeitcDocumento20 pagineReinforcing Steel Placement Handbook: NeitcVel MuruganNessuna valutazione finora
- Das T. and Teng B. 2001 Trust Control and Risk in Strategic Alliances An Integrated Framework'Documento35 pagineDas T. and Teng B. 2001 Trust Control and Risk in Strategic Alliances An Integrated Framework'Vel MuruganNessuna valutazione finora
- RMS DPI 2010-1-49-1: Abdallah N, Liegeois J.P., de Waele B., Fezza N. Et Ouabadi A. The Temaguessine FeDocumento1 paginaRMS DPI 2010-1-49-1: Abdallah N, Liegeois J.P., de Waele B., Fezza N. Et Ouabadi A. The Temaguessine FeVel MuruganNessuna valutazione finora
- Infosecuritystandards 2009 12 FinalDocumento20 pagineInfosecuritystandards 2009 12 FinalVel MuruganNessuna valutazione finora
- Service Manual: Purewatercooler Model Pwc-500/1000/1010/1500Documento38 pagineService Manual: Purewatercooler Model Pwc-500/1000/1010/1500Vel MuruganNessuna valutazione finora
- Vitc Fase Movil AcetonitriloDocumento7 pagineVitc Fase Movil AcetonitriloDaniel Arranz ParaísoNessuna valutazione finora
- Ion Pair ChromatographyDocumento9 pagineIon Pair ChromatographyLeo EspositoNessuna valutazione finora
- 대한민국약전포럼 (Vol 17,+No 1)Documento96 pagine대한민국약전포럼 (Vol 17,+No 1)lichenresearchNessuna valutazione finora
- Glifosate Determination Uv, Por LCDocumento9 pagineGlifosate Determination Uv, Por LCJeiLucasQNessuna valutazione finora
- Chromatography of Aroma Compounds and FragrancesDocumento47 pagineChromatography of Aroma Compounds and Fragrancesilab6638Nessuna valutazione finora
- Tocopherol Concentrate, MixedDocumento4 pagineTocopherol Concentrate, MixedBen ClarkeNessuna valutazione finora
- Jce-1983-393 - Modernization of The Van Deemter EquationDocumento6 pagineJce-1983-393 - Modernization of The Van Deemter EquationSolomon EricksonNessuna valutazione finora
- Biochemistry ResumeDocumento6 pagineBiochemistry Resumeepcbytwhf100% (1)
- CyanocobalaminDocumento2 pagineCyanocobalamingrace_febiantyNessuna valutazione finora
- Rapid Method For Determination of Dehydro Abietic Acid in Gum Rosin and Disproportionate Rosin by Proton Nuclear Magnetic Resonance SpectrosDocumento8 pagineRapid Method For Determination of Dehydro Abietic Acid in Gum Rosin and Disproportionate Rosin by Proton Nuclear Magnetic Resonance SpectrosRian Pratama AkbaNessuna valutazione finora
- 263-Article Text-921-2-10-20190727Documento9 pagine263-Article Text-921-2-10-20190727ankit AcharyaNessuna valutazione finora
- Manual Integration PDFDocumento43 pagineManual Integration PDFnetelsrt1298Nessuna valutazione finora
- Powerpoint Slides - Separation TechniquesDocumento66 paginePowerpoint Slides - Separation Techniquesapi-30590932560% (5)
- Acetogenins From Annonaceae Recent Progress in Isolation Synthesisand Mechanisms of ActionDocumento35 pagineAcetogenins From Annonaceae Recent Progress in Isolation Synthesisand Mechanisms of ActionPanda MndzNessuna valutazione finora
- Lemongrass Oil As A Green PesticideDocumento24 pagineLemongrass Oil As A Green PesticideteguhwidiartoNessuna valutazione finora
- U2103660 - Sharvani - Experiment 6Documento14 pagineU2103660 - Sharvani - Experiment 6SharvaniNessuna valutazione finora
- Pharmacognosy (Effective From The Session - 2016-17)Documento16 paginePharmacognosy (Effective From The Session - 2016-17)fadli_nugraha6109Nessuna valutazione finora
- BiotechniquesDocumento17 pagineBiotechniquesPoypoy Postrano Rojo-ArongNessuna valutazione finora
- USP-NF Cabergoline TabletsDocumento4 pagineUSP-NF Cabergoline Tabletsanon_993394650Nessuna valutazione finora
- TOPIC 15E Spectroscopy and ChromatographyDocumento30 pagineTOPIC 15E Spectroscopy and ChromatographyAyshath MaaishaNessuna valutazione finora
- USP Column ClassificationDocumento9 pagineUSP Column ClassificationKhushbakht KhushiNessuna valutazione finora
- Assignment 2021Documento3 pagineAssignment 2021HemaNessuna valutazione finora
- Jurnal NipaginDocumento6 pagineJurnal NipaginNadya ViraNessuna valutazione finora
- LAYAGUE - Performance Task #1 Paper ChromatographyDocumento3 pagineLAYAGUE - Performance Task #1 Paper ChromatographyNeil LayagueNessuna valutazione finora
- DSE Chemistry - Paper 2 by Dr. Samuel ChongDocumento11 pagineDSE Chemistry - Paper 2 by Dr. Samuel Chonglht001023Nessuna valutazione finora
- Flavor Exploring Citrus Diversity To Create The Next Generation of Citrus FlavorsDocumento9 pagineFlavor Exploring Citrus Diversity To Create The Next Generation of Citrus FlavorsmlankinNessuna valutazione finora
- Chromatography ReviewerDocumento4 pagineChromatography ReviewerAnthony Val RolunaNessuna valutazione finora
- Biological Tools TechniquesDocumento10 pagineBiological Tools Techniquesayushi75086Nessuna valutazione finora
- Dani - An156 - RgaDocumento6 pagineDani - An156 - RgaandreililioanceaNessuna valutazione finora
- Organic Chemistry Some Basic Principles and TechniquesDocumento33 pagineOrganic Chemistry Some Basic Principles and TechniquesAditya Jalal100% (2)