Potrebbero piacerti anche
- Practica Nro 3 Determinacion de Glicemia y Glucosuria BioquimicaDocumento11 paginePractica Nro 3 Determinacion de Glicemia y Glucosuria BioquimicaKELLYNessuna valutazione finora
- Ensayo de Proteínas BradfordDocumento4 pagineEnsayo de Proteínas BradfordJorge Alexander Silva SayagoNessuna valutazione finora
- Clastridium Botulinum - Botulismo Alimenticio en HumanosDocumento15 pagineClastridium Botulinum - Botulismo Alimenticio en HumanosCamila Rios RamosNessuna valutazione finora
- Casos de Episodio Convulsivo y AnsiedadDocumento6 pagineCasos de Episodio Convulsivo y AnsiedadDanielPerezNessuna valutazione finora
- CRUCIGRAMADocumento11 pagineCRUCIGRAMAandyNessuna valutazione finora
- Historia Natural de La Enfermedad de Neumonitis Ocasionada Por Polvos - Output PDFDocumento3 pagineHistoria Natural de La Enfermedad de Neumonitis Ocasionada Por Polvos - Output PDFchristian ruizNessuna valutazione finora
- FT Glucosa en Orina Benedict1Documento2 pagineFT Glucosa en Orina Benedict1Alexander Gelves ParadaNessuna valutazione finora
- Activación de Linfocitos T AlorreactivosDocumento9 pagineActivación de Linfocitos T AlorreactivosManuel Rojas GonzaalesNessuna valutazione finora
- Practica 3: Microbiología III UNIDADDocumento47 paginePractica 3: Microbiología III UNIDADRodolfo Castro100% (1)
- Clostridium TetaniDocumento12 pagineClostridium TetaniElike Pig AttilusNessuna valutazione finora
- Medios CromogenicosDocumento17 pagineMedios CromogenicosMarcoAntonioPalosNessuna valutazione finora
- Test de Antiglobulinas (Test de Combs)Documento68 pagineTest de Antiglobulinas (Test de Combs)MARY100% (2)
- Importancia Del Seguimiento de Los Niveles Séricos de AntiepilépticosDocumento34 pagineImportancia Del Seguimiento de Los Niveles Séricos de AntiepilépticosChristian Escalante Delgado100% (1)
- Caso Hepatitis 3-2021Documento5 pagineCaso Hepatitis 3-2021elisael yinyerNessuna valutazione finora
- Practicas FHDocumento44 paginePracticas FHAlejandra Ceja Vallejo100% (1)
- Banco de Preguntas GeneticaDocumento6 pagineBanco de Preguntas GeneticasamhuhNessuna valutazione finora
- Electroforesis en HemoglobinaDocumento20 pagineElectroforesis en HemoglobinaDiana Cristina Espinoza DuranNessuna valutazione finora
- 7AUTOINMUNIDADDocumento38 pagine7AUTOINMUNIDADVaquero Soleto Maria ReneéNessuna valutazione finora
- Sintomas BucofaringeosDocumento33 pagineSintomas BucofaringeosmarianNessuna valutazione finora
- Microbiología, Patogénesis, Epidemiología, Clínica y Diagnóstico de Las Infecciones Producidas PorDocumento15 pagineMicrobiología, Patogénesis, Epidemiología, Clínica y Diagnóstico de Las Infecciones Producidas PorluisNessuna valutazione finora
- Alteraciones de La Serie BlancaDocumento2 pagineAlteraciones de La Serie BlancaCristian Erubiel Sanchez Lira100% (2)
- CASO CLÍNICO N - 2 Amebas de Vida LibreDocumento2 pagineCASO CLÍNICO N - 2 Amebas de Vida LibreEliel TraucoNessuna valutazione finora
- ANTIGENOSDocumento20 pagineANTIGENOSMarco Antonio Zarate ArceNessuna valutazione finora
- Patologías EritrocitariasDocumento7 paginePatologías EritrocitariasÁlvaroNessuna valutazione finora
- Resumen Principios TP TPT InrDocumento4 pagineResumen Principios TP TPT InrLópez Giraldo YesseniaNessuna valutazione finora
- Práctica 7 Procesos Físicos y Químicos de La DigestiónnnDocumento14 paginePráctica 7 Procesos Físicos y Químicos de La DigestiónnnEthan Ricardo Ramírez AmadorNessuna valutazione finora
- Bacterias de Importancia MedicaDocumento32 pagineBacterias de Importancia MedicaMilagros Quispe Bruno67% (3)
- Genetica Informe 4 Fragilidad CromosomicaDocumento9 pagineGenetica Informe 4 Fragilidad CromosomicaAnthony Ismael Nùñez EstrellaNessuna valutazione finora
- Streptococcus PyogenesDocumento29 pagineStreptococcus PyogenesErick Gerardo EchegarayNessuna valutazione finora
- Pruebas de SensibilidadDocumento10 paginePruebas de SensibilidadDiego Andrés LassoNessuna valutazione finora
- 8 ReporteDocumento5 pagine8 ReporteEliezer Galiego JeronimoNessuna valutazione finora
- Examen MicrobiologiaDocumento5 pagineExamen Microbiologiacindy pumaderaNessuna valutazione finora
- 1 Proteina C ReactivaDocumento8 pagine1 Proteina C ReactivaYasmani MendozaNessuna valutazione finora
- Practica 2-b VDRLDocumento7 paginePractica 2-b VDRLAna Gabriela Fernandez MontenegroNessuna valutazione finora
- Agammaglobulinemia Ligada Al Cromosoma - Jostin CarranzaDocumento5 pagineAgammaglobulinemia Ligada Al Cromosoma - Jostin CarranzaJostin CarranzaNessuna valutazione finora
- Interferon EsDocumento11 pagineInterferon EsEsmeralda LeónNessuna valutazione finora
- Cap 21. Inmunodeficiencias Congénitas y AdquiridasDocumento37 pagineCap 21. Inmunodeficiencias Congénitas y AdquiridasMeliza MoralesNessuna valutazione finora
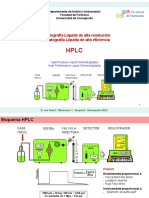
- Problemas HPLCDocumento44 pagineProblemas HPLCLaura Marcela100% (1)
- ZIELHDocumento1 paginaZIELHMarco LojaNessuna valutazione finora
- Caldo BhiDocumento2 pagineCaldo BhiLeonie Neumann GironNessuna valutazione finora
- Sistema Miniturizado API-biomerieuxDocumento5 pagineSistema Miniturizado API-biomerieuxchachito33Nessuna valutazione finora
- Informe de Fagocitosis in VitroDocumento3 pagineInforme de Fagocitosis in VitroJhon Santibañez CalcinaNessuna valutazione finora
- Patología de La Serie Roja 1Documento29 paginePatología de La Serie Roja 1Blas NietoNessuna valutazione finora
- EnterobacteriasDocumento3 pagineEnterobacteriasNESTOR YESID JIMENEZ AFANADORNessuna valutazione finora
- Actividad 4 Histologia ChidaDocumento5 pagineActividad 4 Histologia ChidaAlejandra Luna RuanoNessuna valutazione finora
- Clase 1 Historia de La Inmunologia - Introducción Al S.IDocumento46 pagineClase 1 Historia de La Inmunologia - Introducción Al S.ILuis SilvaNessuna valutazione finora
- FR-Latex: Determinación Cualitativa de Factores Reumatoides (FR)Documento1 paginaFR-Latex: Determinación Cualitativa de Factores Reumatoides (FR)yolo0% (1)
- Clase DX Serologico Brucelosis 2015Documento68 pagineClase DX Serologico Brucelosis 2015ehloco30Nessuna valutazione finora
- Clase HPLC 1 Introducc, F. Móvil, Bombas, InyectorDocumento13 pagineClase HPLC 1 Introducc, F. Móvil, Bombas, Inyectorjuan carlos macaya de la hoz100% (1)
- Practica #3 FisiologíaDocumento5 paginePractica #3 FisiologíaBetun BetyNessuna valutazione finora
- Generalidades GeneticaDocumento3 pagineGeneralidades GeneticaDra Daniela TorresNessuna valutazione finora
- EnterobiasisDocumento10 pagineEnterobiasisJosé Manuel VargasNessuna valutazione finora
- Patogenicidad de Escherichia ColiDocumento9 paginePatogenicidad de Escherichia ColiLeonardoEscalanteNessuna valutazione finora
- Cestode SDocumento26 pagineCestode SdannymalcacamposNessuna valutazione finora
- Western BlotDocumento24 pagineWestern BlotJahuey UnalescoNessuna valutazione finora
- 13 .-EnterobacteriaceaeDocumento21 pagine13 .-EnterobacteriaceaebiotechdogoNessuna valutazione finora
- Leucopenias y Proliferaciones Reactivas de Los Leucocitos y Ganglios LinfaticosDocumento32 pagineLeucopenias y Proliferaciones Reactivas de Los Leucocitos y Ganglios LinfaticosJackelina OrtegaNessuna valutazione finora
- Preguntas ComplementariasDocumento2 paginePreguntas ComplementariasAnonymous TJaFMJNessuna valutazione finora
- InmonologiaDocumento36 pagineInmonologiaOsvaldo SantiagoNessuna valutazione finora
- Sistema Mayor de HistocompatibilidadDocumento28 pagineSistema Mayor de HistocompatibilidadCarlosJuniorCalleNessuna valutazione finora
- Evidence-Based Concepts and - En.esDocumento19 pagineEvidence-Based Concepts and - En.esCristian FernandoNessuna valutazione finora
- Aula Virtual - CLASIF HI, MICRO LeSage 2007 - Aesthetic Anterior Composite Restorations - A Guide To Direct Placement - En.esDocumento20 pagineAula Virtual - CLASIF HI, MICRO LeSage 2007 - Aesthetic Anterior Composite Restorations - A Guide To Direct Placement - En.esCristian FernandoNessuna valutazione finora
- LIBRO Odontologia - Tratado de Cirugia Bucal - Tomo I - Cosme GayDocumento850 pagineLIBRO Odontologia - Tratado de Cirugia Bucal - Tomo I - Cosme GayMaria Fernanda Jorquera Campe96% (475)
- Effect of Access Cavity Location and Design On Degree and Distribution of Instrumented Root Canal Surface in Maxillary Anterior Teeth Mannan2001.en - Es PDFDocumento8 pagineEffect of Access Cavity Location and Design On Degree and Distribution of Instrumented Root Canal Surface in Maxillary Anterior Teeth Mannan2001.en - Es PDFCristian FernandoNessuna valutazione finora
- Comparison of Apical Centring Ability Between Incisal-Shifted Access and Traditional Lingual Access For Maxillary Anterior Teeth - En.es PDFDocumento6 pagineComparison of Apical Centring Ability Between Incisal-Shifted Access and Traditional Lingual Access For Maxillary Anterior Teeth - En.es PDFCristian FernandoNessuna valutazione finora
- Labial Access Opening in Mandibular Anterior Teethâ - An Alternative Approach To Success - En.es PDFDocumento7 pagineLabial Access Opening in Mandibular Anterior Teethâ - An Alternative Approach To Success - En.es PDFCristian FernandoNessuna valutazione finora
- Ficha Clinica de PeriodonciaDocumento6 pagineFicha Clinica de PeriodonciaCristian FernandoNessuna valutazione finora
- 36-37 Replicacion TranscripcionDocumento14 pagine36-37 Replicacion TranscripcionCristian FernandoNessuna valutazione finora
- Cris ExpoDocumento33 pagineCris ExpoCristian FernandoNessuna valutazione finora
- Tratamiento Farmacológico Pre y Post Exodoncia: Aplicación de La DexametasonaDocumento13 pagineTratamiento Farmacológico Pre y Post Exodoncia: Aplicación de La DexametasonaCristian FernandoNessuna valutazione finora
- 11 LenguaDocumento34 pagine11 LenguaCristian FernandoNessuna valutazione finora
- Protesis Fija y RemovibleDocumento47 pagineProtesis Fija y RemovibleCristian FernandoNessuna valutazione finora
- El Zinc El La Salud Del CabelloDocumento11 pagineEl Zinc El La Salud Del Cabellooreaj2Nessuna valutazione finora
- Trastornos RespiratoriosDocumento19 pagineTrastornos RespiratoriosLI CorazoncitoNessuna valutazione finora
- Norma para El Manejo Terapeutico de Las Personas Con Vih en La Republica de PanamaDocumento137 pagineNorma para El Manejo Terapeutico de Las Personas Con Vih en La Republica de PanamaDavid Montalván HurtadoNessuna valutazione finora
- Unidad-II-tema-1 - Errores Innatos Del MetabolismoDocumento55 pagineUnidad-II-tema-1 - Errores Innatos Del MetabolismoKenett SerranoNessuna valutazione finora
- Fichas Tecnicas De, Microorganismos de Importancia en Microbiología.Documento16 pagineFichas Tecnicas De, Microorganismos de Importancia en Microbiología.karla janeth cruz escobarNessuna valutazione finora
- Fito FormulasDocumento38 pagineFito FormulasEn La Luna LuneraNessuna valutazione finora
- Cigarro Doc 1Documento5 pagineCigarro Doc 1Emerson Pacheco RiveroNessuna valutazione finora
- Mapa Conceptual Vaso Dilatadores-1Documento7 pagineMapa Conceptual Vaso Dilatadores-1Andrea Carolina Baron HurtadoNessuna valutazione finora
- Tratamiento y Características de Trastorno de PersonalidadDocumento9 pagineTratamiento y Características de Trastorno de PersonalidadYT DebestiaNessuna valutazione finora
- Introducción AterosclerosisDocumento2 pagineIntroducción AterosclerosisVanessa CaltranNessuna valutazione finora
- 1.3 Interpretación de Las Puntuaciones EstandarizadasDocumento4 pagine1.3 Interpretación de Las Puntuaciones EstandarizadasClara GordanoNessuna valutazione finora
- RetroviridaeDocumento8 pagineRetroviridaeMaría QuispeNessuna valutazione finora
- ¿Qué Son?: NecrosisDocumento2 pagine¿Qué Son?: NecrosiskikilinNessuna valutazione finora
- Plan de Higiene Ocupacional - 2024-2025Documento12 paginePlan de Higiene Ocupacional - 2024-2025Kum Gustavo MamaniNessuna valutazione finora
- Semiologia Medica 3a Edicion Capítulo 26 y 27Documento11 pagineSemiologia Medica 3a Edicion Capítulo 26 y 27Cinthia Beatriz Valenzuela MendezNessuna valutazione finora
- Construcciones en AlturasDocumento5 pagineConstrucciones en AlturasJaimito SalazarNessuna valutazione finora
- Alfabeto GriegoDocumento16 pagineAlfabeto GriegoAlddo Resèndiz100% (1)
- Electiva Marby Gomez 30338696Documento9 pagineElectiva Marby Gomez 30338696Marby GomezNessuna valutazione finora
- MÓDULO II EleudimarDocumento7 pagineMÓDULO II EleudimarEleudimarNessuna valutazione finora
- Capitulo I. Curi Giron, Sadith Mercedes Giraldez Moran, Karol NinfaDocumento10 pagineCapitulo I. Curi Giron, Sadith Mercedes Giraldez Moran, Karol NinfaJose VillanuevaNessuna valutazione finora
- Fundamentos de Cirugia Laparoscopica 1Documento72 pagineFundamentos de Cirugia Laparoscopica 1CesarCisneros100% (1)
- Anemia GeneralDocumento2 pagineAnemia GeneralChristine GarciaNessuna valutazione finora
- Victor Vera Cisternas - Tarea 6 - Proyecto de TituloDocumento8 pagineVictor Vera Cisternas - Tarea 6 - Proyecto de TituloVictorVeraCisterna0% (1)
- Calendario Cdcperu 2023 PDFDocumento1 paginaCalendario Cdcperu 2023 PDFMaCley Abundo TrujilloNessuna valutazione finora
- FICHA TECNICA Patty OxacilinaDocumento16 pagineFICHA TECNICA Patty OxacilinaMauricio Orlando Vanegas CasadiegoNessuna valutazione finora
- Ifu Dialog Iq Esrev10401Documento442 pagineIfu Dialog Iq Esrev10401FELIPE ANDRESNessuna valutazione finora
- Carcinoma de Mama en Trabajadora Expuesta A Radiaciones IonizantesDocumento7 pagineCarcinoma de Mama en Trabajadora Expuesta A Radiaciones IonizantesElizabeth Karine Herrera100% (1)
- Tipos de BrigadasDocumento2 pagineTipos de Brigadasjguerrero2350% (12)
- GastritisDocumento4 pagineGastritisYeny CeballosNessuna valutazione finora
- Informe de La Práctica Huevos de Enterobius VermicularisDocumento6 pagineInforme de La Práctica Huevos de Enterobius VermicularisJESUS ANDREA BRINGAS DE LA CRUZNessuna valutazione finora