Potrebbero piacerti anche
- MONOGRAFIADocumento18 pagineMONOGRAFIAtanyNessuna valutazione finora
- E Stereo Qui MicaDocumento35 pagineE Stereo Qui MicaAngeles RojosNessuna valutazione finora
- Presentación Final Equipo 1Documento22 paginePresentación Final Equipo 1Karla DelgadoNessuna valutazione finora
- 13 Isomeriaoptica 2011Documento21 pagine13 Isomeriaoptica 2011Lorena RojasNessuna valutazione finora
- Conf. No.5 ESTEREOQUIMICADocumento58 pagineConf. No.5 ESTEREOQUIMICAJeny Serrano0% (1)
- EstereoquimicaDocumento65 pagineEstereoquimicaminionmaze1Nessuna valutazione finora
- Enantiómeros: Presentado A: Yurany CamachoDocumento15 pagineEnantiómeros: Presentado A: Yurany Camachocamila cristanchoNessuna valutazione finora
- ESTEREOQUIMICADocumento11 pagineESTEREOQUIMICAAnny ChaustreNessuna valutazione finora
- Antología de Química Orgánica IDocumento44 pagineAntología de Química Orgánica ILunaNessuna valutazione finora
- Práctica de EstereoquímicaDocumento8 paginePráctica de EstereoquímicaYordan SantanaNessuna valutazione finora
- IsomeríaDocumento21 pagineIsomeríaЧydr0geИ ТоварищNessuna valutazione finora
- Esteroquimica FinalDocumento23 pagineEsteroquimica FinalErik Rosmel Ordoñez AlcoserNessuna valutazione finora
- Introduccion de QuimicaDocumento22 pagineIntroduccion de QuimicaVladimir Huapaya ChavezNessuna valutazione finora
- EstereoquimicaDocumento7 pagineEstereoquimicaYovana RojasNessuna valutazione finora
- Diferencia Entre Enantiometos y DiasteromerosDocumento10 pagineDiferencia Entre Enantiometos y Diasteromeroscarlos vallejoNessuna valutazione finora
- Quiral PutoDocumento34 pagineQuiral Putobarbara potulniskyNessuna valutazione finora
- Trabajo Final Orgánica 1Documento23 pagineTrabajo Final Orgánica 1Jesús MorenoNessuna valutazione finora
- Práctica 9 EsterioquímicaDocumento12 paginePráctica 9 EsterioquímicaValentin John ApazaNessuna valutazione finora
- Química Orgánica. Estereoquímica.Documento14 pagineQuímica Orgánica. Estereoquímica.Pablo Miguel González JuddNessuna valutazione finora
- Informe QuímicaDocumento37 pagineInforme QuímicaCamila LópezNessuna valutazione finora
- Quimica Organica I 05 EstereoquimicaDocumento88 pagineQuimica Organica I 05 EstereoquimicaAlex Ramos maqueraNessuna valutazione finora
- Informe QuímicaDocumento37 pagineInforme QuímicaCamila LópezNessuna valutazione finora
- EstereoquimicaDocumento11 pagineEstereoquimicaSantiago ZabalaNessuna valutazione finora
- Tema 4 IsomeriaDocumento28 pagineTema 4 IsomeriaCHAVEZ RUIZ DAYANANessuna valutazione finora
- Tarea AC05 ConfiguraciónDocumento5 pagineTarea AC05 ConfiguraciónJESSICA GUADALUPE BANUELOS JIMENEZNessuna valutazione finora
- INFOREME - EstereoquimicaDocumento15 pagineINFOREME - EstereoquimicaDiego Raúl Vásquez TelloNessuna valutazione finora
- La IsomeriaDocumento14 pagineLa IsomeriamariselaNessuna valutazione finora
- Guia IsomerosDocumento7 pagineGuia IsomerosValeria ApablazaNessuna valutazione finora
- Unidad III. Isomería.Documento18 pagineUnidad III. Isomería.Janet Paulina Ramirez MoralesNessuna valutazione finora
- HomoquiralidadDocumento16 pagineHomoquiralidadEllen AguilarNessuna valutazione finora
- Trabajo de Isomeria Cis TransDocumento28 pagineTrabajo de Isomeria Cis TransKaren Aguillon VeraNessuna valutazione finora
- Tarea 2 FaltaDocumento15 pagineTarea 2 FaltaEdson LaricoNessuna valutazione finora
- Trabajo Monografico 1Documento10 pagineTrabajo Monografico 1Franco Garcia ValeraNessuna valutazione finora
- Informe 2-Alania-Pinto Quim. OrgánicaDocumento24 pagineInforme 2-Alania-Pinto Quim. OrgánicaMilagros AleNessuna valutazione finora
- Unidad 4. ESTEREOISOMERÍADocumento21 pagineUnidad 4. ESTEREOISOMERÍANeida MedinaNessuna valutazione finora
- Informe 4 EstereoisomeriaDocumento9 pagineInforme 4 EstereoisomeriaLuis Alberto Manrique PenagosNessuna valutazione finora
- EstereoquímicaDocumento3 pagineEstereoquímicaRicardo García ANessuna valutazione finora
- EstereoquímicaDocumento6 pagineEstereoquímicalcgaNessuna valutazione finora
- IsomeríaDocumento10 pagineIsomeríaAimméMirandaMoralesNessuna valutazione finora
- Presentación 4-EstereoquímicaDocumento52 paginePresentación 4-EstereoquímicaLINA MARIA ORTIZ ULLOANessuna valutazione finora
- Estereoquimica e IsomeriaDocumento3 pagineEstereoquimica e IsomeriaKayvaan ShrikeNessuna valutazione finora
- Franye 1Documento8 pagineFranye 1Duban HernandezNessuna valutazione finora
- Estereoquimica de Los PolimerosDocumento4 pagineEstereoquimica de Los PolimerosJavier Heredia GodoyNessuna valutazione finora
- IsomerosDocumento10 pagineIsomerosLuis AlfonsoNessuna valutazione finora
- Actividad 1 Und 5Documento6 pagineActividad 1 Und 50102 Alejandro Alvarez LunaNessuna valutazione finora
- IsómerosDocumento4 pagineIsómerosSergio YepezNessuna valutazione finora
- Lab1 (Modelos Moleculares)Documento16 pagineLab1 (Modelos Moleculares)Arturo Mansilla JordanNessuna valutazione finora
- 3.1.-Cromatrografia Des. Hist.Documento30 pagine3.1.-Cromatrografia Des. Hist.ALANNessuna valutazione finora
- Separaciones Quimicas y EnantiomerismoDocumento29 pagineSeparaciones Quimicas y EnantiomerismoJavier RuizNessuna valutazione finora
- EstereoquímicaDocumento30 pagineEstereoquímicaAna GuzmánNessuna valutazione finora
- Clase 7. Tema IIDocumento33 pagineClase 7. Tema IIRolleros unidosNessuna valutazione finora
- Monografia MikeDocumento21 pagineMonografia MikeMike GonzalesNessuna valutazione finora
- Esteroisomería y EstereoisómerosDocumento15 pagineEsteroisomería y EstereoisómerosLaura ColomNessuna valutazione finora
- Estereoquimica Doc Oficial-1Documento17 pagineEstereoquimica Doc Oficial-1Leticia NavíaNessuna valutazione finora
- EstereoquímicaDocumento17 pagineEstereoquímicarmarin_90Nessuna valutazione finora
- Introducción al mundo cuántico: De la danza de las partículas a las semillas de las galaxiasDa EverandIntroducción al mundo cuántico: De la danza de las partículas a las semillas de las galaxiasNessuna valutazione finora
- Hibridación de Los Ácidos NucleicosDocumento1 paginaHibridación de Los Ácidos NucleicosMons JassoNessuna valutazione finora
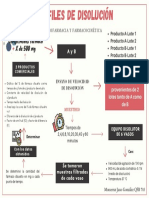
- Diagrama de Flujo PerfilDocumento1 paginaDiagrama de Flujo PerfilMons JassoNessuna valutazione finora
- Ácidos NucleicosDocumento2 pagineÁcidos NucleicosMons JassoNessuna valutazione finora
- Análisis BioinfoDocumento7 pagineAnálisis BioinfoMons JassoNessuna valutazione finora
- Textos Foto DedicatoriaDocumento1 paginaTextos Foto DedicatoriaMons JassoNessuna valutazione finora
- Búsqueda Bibliográfica en PubMedDocumento4 pagineBúsqueda Bibliográfica en PubMedMons JassoNessuna valutazione finora
- MATRIZ - Monserrat Jasso GonzálezDocumento2 pagineMATRIZ - Monserrat Jasso GonzálezMons JassoNessuna valutazione finora
- Mapa Mental - Desarrollo de Nuevos API S PDFDocumento1 paginaMapa Mental - Desarrollo de Nuevos API S PDFMons JassoNessuna valutazione finora
- VirusDocumento21 pagineVirusMons JassoNessuna valutazione finora
- GentamicinaDocumento3 pagineGentamicinaMons JassoNessuna valutazione finora
- Gráfico Del Porcentaje de Fármaco Disuelto Contra Tiempo de Cada Lote de Cada Producto en EstudioDocumento18 pagineGráfico Del Porcentaje de Fármaco Disuelto Contra Tiempo de Cada Lote de Cada Producto en EstudioMons JassoNessuna valutazione finora
- AzitromicinaDocumento3 pagineAzitromicinaMons JassoNessuna valutazione finora
- CiprofloxacinoDocumento6 pagineCiprofloxacinoMons JassoNessuna valutazione finora
- G ProteinDocumento5 pagineG ProteinMons Jasso100% (1)
- Gráfico Del Porcentaje de Fármaco Disuelto Contra Tiempo de Cada Lote de Cada Producto en EstudioDocumento18 pagineGráfico Del Porcentaje de Fármaco Disuelto Contra Tiempo de Cada Lote de Cada Producto en EstudioMons JassoNessuna valutazione finora
- Pres2 EquipoDocumento1 paginaPres2 EquipoPatricia Morales LinaresNessuna valutazione finora
- Pres2 EquipoDocumento1 paginaPres2 EquipoPatricia Morales LinaresNessuna valutazione finora
- Checklist-TECNO 2Documento1 paginaChecklist-TECNO 2Mons JassoNessuna valutazione finora
- Metodología de La Investigación - ActiDocumento6 pagineMetodología de La Investigación - ActiMons JassoNessuna valutazione finora
- MorfofisiologiaDocumento6 pagineMorfofisiologiaMons JassoNessuna valutazione finora
- Libro Principios de EticaDocumento294 pagineLibro Principios de EticaShere Khan100% (5)
- Pres4 IndividualDocumento3 paginePres4 IndividualMons JassoNessuna valutazione finora
- Plan de Mejora Q.F.B. 2015 PDFDocumento46 paginePlan de Mejora Q.F.B. 2015 PDFMons JassoNessuna valutazione finora
- Trabajo Célula EucariotaDocumento11 pagineTrabajo Célula EucariotaMons JassoNessuna valutazione finora
- Práctica No. 1Documento5 paginePráctica No. 1Mons JassoNessuna valutazione finora
- Practica 1 Anatomia Macroscopica de Los Sistemas de OrganosDocumento9 paginePractica 1 Anatomia Macroscopica de Los Sistemas de OrganosMons JassoNessuna valutazione finora
- Práctica 1 Anatomía Macroscópica de Los Sistemas de ÓrganosDocumento15 paginePráctica 1 Anatomía Macroscópica de Los Sistemas de Órganosthecachi100% (1)
- Practica de Estructura Quimica 3ro SecundariaDocumento3 paginePractica de Estructura Quimica 3ro SecundariaJorge Arcadio100% (1)
- Sílabo Matemática Básica MFeIDocumento7 pagineSílabo Matemática Básica MFeIcesar vasquez trejoNessuna valutazione finora
- Exposición Cibernetica - Pensamiento SistemicoDocumento41 pagineExposición Cibernetica - Pensamiento SistemicoBrayan VargasNessuna valutazione finora
- Laboratorio de Yacimientos Talara PDFDocumento66 pagineLaboratorio de Yacimientos Talara PDFNilo Llamocca GutierrezNessuna valutazione finora
- Geometría III Actividad IDocumento5 pagineGeometría III Actividad IJulio Cesar RodriguezNessuna valutazione finora
- Ec (1) (045-049) .En - EsDocumento5 pagineEc (1) (045-049) .En - EsDavid AlanocaNessuna valutazione finora
- 3.5.-Operacion y Cuidados CribasDocumento68 pagine3.5.-Operacion y Cuidados CribasFrancisco Meza GonzalezNessuna valutazione finora
- Repartido N°2 - Campo EléctricoDocumento2 pagineRepartido N°2 - Campo EléctricoJennylorena Calo amorinNessuna valutazione finora
- Concentración JigsDocumento20 pagineConcentración Jigsberniegd100% (1)
- MatematicasDocumento9 pagineMatematicasmarly rodriguezNessuna valutazione finora
- Radiación de HawkingDocumento4 pagineRadiación de HawkingomarNessuna valutazione finora
- SOPLADODocumento42 pagineSOPLADOJacques Robert Gonzalez100% (3)
- Preparación de SolucionesDocumento30 paginePreparación de SolucionesLuis Arturo Ruiz Galindo100% (2)
- UniversoDocumento17 pagineUniversoJhon Lap LeOo EsStonner PonceNessuna valutazione finora
- Transformadores de ProteccionDocumento34 pagineTransformadores de ProteccionLisandroNessuna valutazione finora
- Tipo de Actuadores EléctricosDocumento4 pagineTipo de Actuadores EléctricosEnrique AlvarezNessuna valutazione finora
- 7.2.1 Divisibilidad y Numeros Primos y CompuestosDocumento8 pagine7.2.1 Divisibilidad y Numeros Primos y Compuestoslianeuse100% (2)
- 0348-ESP-AEE-000C-000-0007 KKS Codificación de KKS de Equipos y Componentes PDFDocumento87 pagine0348-ESP-AEE-000C-000-0007 KKS Codificación de KKS de Equipos y Componentes PDFObedJiménez100% (1)
- Problemas Del Tema de EnergíasDocumento3 pagineProblemas Del Tema de EnergíasArqus1Nessuna valutazione finora
- H 50 N 01 A 571 NLNJT 38 Tksllne 216 LMBDocumento4 pagineH 50 N 01 A 571 NLNJT 38 Tksllne 216 LMBWinny leslyNessuna valutazione finora
- Previo 2 LemDocumento7 paginePrevio 2 LemSandro ReyesNessuna valutazione finora
- Metodos Kani Taka y Otros PDFDocumento44 pagineMetodos Kani Taka y Otros PDFYessenia ElisaNessuna valutazione finora
- Central Termica IloDocumento1 paginaCentral Termica IloDulcemariaAlvarescNessuna valutazione finora
- Unidad Temática #13 PagDocumento20 pagineUnidad Temática #13 PagCarlos VanzanNessuna valutazione finora
- Lab 7 LEY DE OHMDocumento14 pagineLab 7 LEY DE OHMPabloRafaelSotoNessuna valutazione finora
- SPC 3 Ppa7 Trimestre 2Documento15 pagineSPC 3 Ppa7 Trimestre 2Confezor CoderoNessuna valutazione finora
- 14 Ficha TaiChiYang 32 EspadaDocumento18 pagine14 Ficha TaiChiYang 32 EspadaAnonymous rzCuQf100% (1)
- Tarea de Conservacion PiagetDocumento10 pagineTarea de Conservacion PiagetprofexNessuna valutazione finora
- Venegas Ss PDFDocumento126 pagineVenegas Ss PDFKarina MontesNessuna valutazione finora