Potrebbero piacerti anche
- TRAB INVEST Trauma Nasal FinalDocumento21 pagineTRAB INVEST Trauma Nasal Finalvir77saNessuna valutazione finora
- Fisiopatologia de La SepsisDocumento14 pagineFisiopatologia de La SepsisJoakoRuizVillegasNessuna valutazione finora
- Lupus y EmbarazoDocumento11 pagineLupus y EmbarazoFrancisco Javier PerezNessuna valutazione finora
- Derrame pleural: causas, diagnóstico y tratamientoDocumento22 pagineDerrame pleural: causas, diagnóstico y tratamientoSergio JavierNessuna valutazione finora
- Resumen de Antibioticos en OrtopediaDocumento7 pagineResumen de Antibioticos en OrtopediaGómez Agustín Luis FeNessuna valutazione finora
- Leucemias: clasificación, signos y tratamientoDocumento8 pagineLeucemias: clasificación, signos y tratamientoCarlos Astete BórquezNessuna valutazione finora
- Vih y Embarazo AngelaDocumento11 pagineVih y Embarazo AngelaJULIAN DARIO AGUILERA TERCERONessuna valutazione finora
- Caso Clinico de Molluscum ContagiosumDocumento4 pagineCaso Clinico de Molluscum Contagiosumelisa davilaNessuna valutazione finora
- Artritis reumatoide: guía esencialDocumento10 pagineArtritis reumatoide: guía esencialGio MirandaNessuna valutazione finora
- Sindrome Atifosfolipido y EmbarazoDocumento22 pagineSindrome Atifosfolipido y EmbarazoJorge AndradeNessuna valutazione finora
- Examen de GradoDocumento33 pagineExamen de Gradojorgerc1995Nessuna valutazione finora
- HidatidosisDocumento19 pagineHidatidosisPaula SagredoNessuna valutazione finora
- Síndrome de SheehanDocumento3 pagineSíndrome de SheehanLeo ChavezNessuna valutazione finora
- Mordedura Por LoxocelesDocumento9 pagineMordedura Por LoxocelesAndre JailaNessuna valutazione finora
- Neoplasia Intraepitelial CervicalDocumento11 pagineNeoplasia Intraepitelial CervicalElizabeth Mautino CaceresNessuna valutazione finora
- Anemias Pediatria Version FinalDocumento25 pagineAnemias Pediatria Version Finallisita23_182Nessuna valutazione finora
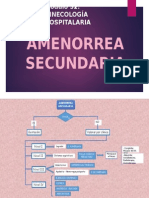
- Amenorrea SecundariaDocumento2 pagineAmenorrea SecundariaLuly Caro ArmasNessuna valutazione finora
- Indicadores de Consulta Externa de Pediatria Tercer NivelDocumento6 pagineIndicadores de Consulta Externa de Pediatria Tercer NivelfragajavierNessuna valutazione finora
- Protocolo Pre QXDocumento6 pagineProtocolo Pre QXWalter Barriga ManriqueNessuna valutazione finora
- 06 SX MieloproliferativosDocumento15 pagine06 SX MieloproliferativosSandy Salazar100% (1)
- Infecciones Respiratorias Agudas en Pediatria Ambulatoria PucDocumento6 pagineInfecciones Respiratorias Agudas en Pediatria Ambulatoria Puclorenitha10Nessuna valutazione finora
- Quiste HidatidicoDocumento8 pagineQuiste HidatidicoAlexis DiosesNessuna valutazione finora
- Quemadura Por CausticosDocumento71 pagineQuemadura Por CausticosSt@r goldenNessuna valutazione finora
- Artropatía Por Cristales Gota PDFDocumento5 pagineArtropatía Por Cristales Gota PDFPaola CFNessuna valutazione finora
- VIH - Dr. JulioDocumento5 pagineVIH - Dr. JulioSOFIA ALEJANDRA SANABRIA FANDIÑONessuna valutazione finora
- Pancreatitis CrónicaDocumento3 paginePancreatitis CrónicaVirginia López BarberoNessuna valutazione finora
- AntibioticosDocumento39 pagineAntibioticosFdo RMNessuna valutazione finora
- Tratamiento de La Infección Por El VIH. Fármacos Antirretrovirales PDFDocumento13 pagineTratamiento de La Infección Por El VIH. Fármacos Antirretrovirales PDFhopeheal100% (1)
- Mieloma MultipleDocumento8 pagineMieloma MultipleJennifer Wever LorenzoNessuna valutazione finora
- Anemia MegaloblasticaDocumento4 pagineAnemia MegaloblasticaFrancis MunguíaNessuna valutazione finora
- Examen de La Mujer GravidaDocumento15 pagineExamen de La Mujer Gravidagarfield_12Nessuna valutazione finora
- 1 Aspectos Legales Primeros AuxiliosDocumento261 pagine1 Aspectos Legales Primeros AuxiliosMILENA SALAMANCANessuna valutazione finora
- Manifestaciones Cutaneas de Enfermedades SistemicasDocumento32 pagineManifestaciones Cutaneas de Enfermedades SistemicasJoys Saucedo SaldivarNessuna valutazione finora
- Apendicitis Aguda en Pediatria CompletoDocumento31 pagineApendicitis Aguda en Pediatria CompletoKelly Watson0% (1)
- EsclerodermiaDocumento36 pagineEsclerodermiaSofhy CastellanoNessuna valutazione finora
- Fisiopatología Del ComaDocumento15 pagineFisiopatología Del Comaale1422Nessuna valutazione finora
- Infecciones Asociadas A La Atención en SaludDocumento4 pagineInfecciones Asociadas A La Atención en SaludJaimeNessuna valutazione finora
- Temario Simplificado IPNDocumento7 pagineTemario Simplificado IPNMOCHABATYNessuna valutazione finora
- Esofagitis Por CáusticosDocumento14 pagineEsofagitis Por CáusticosSara Lucía AchoyNessuna valutazione finora
- Casos ClinicosDocumento36 pagineCasos ClinicosAlejandra MonteroNessuna valutazione finora
- ENCEFALOPATÍA HIPERTENSIVA: CAUSAS, SÍNTOMAS Y DIAGNÓSTICODocumento4 pagineENCEFALOPATÍA HIPERTENSIVA: CAUSAS, SÍNTOMAS Y DIAGNÓSTICOEricksen SalazarNessuna valutazione finora
- Examen de La Mujer GrávidaDocumento21 pagineExamen de La Mujer GrávidaAntonella GómezNessuna valutazione finora
- Síndrome de Anticuerpos Antifosfolípido y EmbarazoDocumento29 pagineSíndrome de Anticuerpos Antifosfolípido y EmbarazomipdanceNessuna valutazione finora
- Manejo de Líquidos y Electrólitos - ResumenDocumento14 pagineManejo de Líquidos y Electrólitos - ResumenPaúl AltamiranoNessuna valutazione finora
- Monografia FINALDocumento13 pagineMonografia FINALjgonzales_323696Nessuna valutazione finora
- Cuerpos Extraños en Via Digestiva Alta, enDocumento33 pagineCuerpos Extraños en Via Digestiva Alta, enVR AlvaroNessuna valutazione finora
- Caso Clinico 2 - Med - InternaDocumento12 pagineCaso Clinico 2 - Med - InternaRaFael LojaNessuna valutazione finora
- PielonefritisDocumento4 paginePielonefritiscateterdoblejotaNessuna valutazione finora
- Fisiopatologia de La SepsisDocumento26 pagineFisiopatologia de La SepsisKelly Franco BustamanteNessuna valutazione finora
- Hipertensión: Factores de riesgo, fisiopatología y prevenciónDocumento14 pagineHipertensión: Factores de riesgo, fisiopatología y prevenciónGenesis BalzaNessuna valutazione finora
- Caso Clínico DengueDocumento9 pagineCaso Clínico DengueALEINADNessuna valutazione finora
- Sepsis y Embarazo Guia FLASOG 2013 PDFDocumento19 pagineSepsis y Embarazo Guia FLASOG 2013 PDFEdwin BravoNessuna valutazione finora
- Clase Sindrome Mieloproliferativos Parte 1Documento48 pagineClase Sindrome Mieloproliferativos Parte 1Tell Me DaryNessuna valutazione finora
- Artritis Reumatoide y EmbarazoDocumento3 pagineArtritis Reumatoide y Embarazoapi-520462920Nessuna valutazione finora
- GonorreaDocumento30 pagineGonorreaAndre MB100% (1)
- Sindrome NefróticoDocumento9 pagineSindrome NefróticoFrancisca RiquelmeNessuna valutazione finora
- Shock Distributivo ResumenDocumento4 pagineShock Distributivo ResumenEvelyn Milagros MamaniNessuna valutazione finora
- Gangrena gaseosa causada por ClostridiumDocumento4 pagineGangrena gaseosa causada por ClostridiumLIZETH DAYANA OCHOA RODRIGUEZ100% (1)
- Fisiopatologia de La SepsisDocumento26 pagineFisiopatologia de La SepsisKim Hyun JoongNessuna valutazione finora
- Actualización en Sepsis y Choque Séptico Nuevas Definiciones y Evaluación Clínica PDFDocumento26 pagineActualización en Sepsis y Choque Séptico Nuevas Definiciones y Evaluación Clínica PDFKoraima Villar Corzo100% (1)
- Primer Examen Cpu 2004-III Grupo III - Tema p1Documento14 paginePrimer Examen Cpu 2004-III Grupo III - Tema p1John Frank Zaldivar Rios67% (3)
- Informe de Prueba PilotoDocumento1 paginaInforme de Prueba PilotoJohn Frank Zaldivar RiosNessuna valutazione finora
- Calidad de Vida en TEC Post Accidente de TransitoDocumento8 pagineCalidad de Vida en TEC Post Accidente de TransitoJohn Frank Zaldivar RiosNessuna valutazione finora
- Examen Admision 2005 OrdinarioDocumento20 pagineExamen Admision 2005 OrdinarioJohn Frank Zaldivar Rios100% (1)
- Tratamiento Osteoporosis 2014Documento10 pagineTratamiento Osteoporosis 2014John Frank Zaldivar RiosNessuna valutazione finora
- Primer Examen Cpu 2005-III Grupo III-tema PDocumento13 paginePrimer Examen Cpu 2005-III Grupo III-tema PJohn Frank Zaldivar Rios100% (2)
- Ordinario 2006-I Completo 149 PregDocumento20 pagineOrdinario 2006-I Completo 149 PregJohn Frank Zaldivar Rios100% (1)
- Egresados 2008 IDocumento13 pagineEgresados 2008 IJohn Frank Zaldivar Rios100% (2)
- Términos de Referencia CiclovíaDocumento2 pagineTérminos de Referencia CiclovíaCarlos Cristian Castro LinaresNessuna valutazione finora
- Taller Reconoce La Química Como Una Herramienta para La Vida 2018Documento14 pagineTaller Reconoce La Química Como Una Herramienta para La Vida 2018Sofia CastilloNessuna valutazione finora
- Post - Tarea - Evaluación FinalDocumento13 paginePost - Tarea - Evaluación FinalArselioRuizPerezNessuna valutazione finora
- Caso Practico Lab QuimicaDocumento2 pagineCaso Practico Lab QuimicaRodolfo Bautista RodriguezNessuna valutazione finora
- Vtec HondaDocumento31 pagineVtec Hondaxshanwmichaelsx100% (2)
- Guía de Asesoría SASSTDocumento17 pagineGuía de Asesoría SASSTraulNessuna valutazione finora
- La Temperatura Del Cuerpo Humano 2Documento3 pagineLa Temperatura Del Cuerpo Humano 2liz isabella garcia duranNessuna valutazione finora
- Evaluación BiológicaDocumento2 pagineEvaluación BiológicaIvonne OrtegaNessuna valutazione finora
- A Mi También Me Duele El Duelo Propio Del AcompañanteDocumento26 pagineA Mi También Me Duele El Duelo Propio Del Acompañantesonia_mata_9Nessuna valutazione finora
- Medicina Legal Trabajo CompletoDocumento26 pagineMedicina Legal Trabajo CompletoSergio CalderónNessuna valutazione finora
- Factores Que Intervienen en El Desarrollo Del Potencial de AcciónDocumento6 pagineFactores Que Intervienen en El Desarrollo Del Potencial de AcciónCecilia PachecoNessuna valutazione finora
- Bognorte E02 3475124 RmcerebroDocumento2 pagineBognorte E02 3475124 RmcerebroTomas LopezNessuna valutazione finora
- 4.-Especificaciones Instalaciones ElectricasDocumento6 pagine4.-Especificaciones Instalaciones ElectricasKevin Cardenas MendozaNessuna valutazione finora
- Estudio Prefactibilidad Caracol....Documento73 pagineEstudio Prefactibilidad Caracol....Antonio PeñaNessuna valutazione finora
- CatalogoPublicaciones2016 PDFDocumento210 pagineCatalogoPublicaciones2016 PDFPascual Bernal OrduñoNessuna valutazione finora
- RFC emisor ZAVAR SOLUTIONS S.A. DE C.VDocumento1 paginaRFC emisor ZAVAR SOLUTIONS S.A. DE C.VFer ZuñigaNessuna valutazione finora
- Causas y tratamiento de la ascitisDocumento6 pagineCausas y tratamiento de la ascitisTobias LaubNessuna valutazione finora
- Taller Repaso Balance de MasaDocumento1 paginaTaller Repaso Balance de Masasilvia olarteNessuna valutazione finora
- Procesos Psicologicos Basicos en El Estudio Del Comportamiento Del ConsumidorDocumento7 pagineProcesos Psicologicos Basicos en El Estudio Del Comportamiento Del ConsumidorCamilo GonzaezNessuna valutazione finora
- Prob #5 Balance de EnergiaDocumento5 pagineProb #5 Balance de EnergiaAdriana Lorena Zenteno TejadaNessuna valutazione finora
- Cod. 0972 - Biologia IiiDocumento32 pagineCod. 0972 - Biologia IiiEDITH YOLANDA Tolosa cuadradoNessuna valutazione finora
- APARTADO III CorregidoDocumento8 pagineAPARTADO III CorregidoEsteban EscuderoNessuna valutazione finora
- Estudio de Caso Baja AutoestimaDocumento29 pagineEstudio de Caso Baja Autoestimabmari17Nessuna valutazione finora
- Estudio de Caso Pedagogía EducativaDocumento2 pagineEstudio de Caso Pedagogía EducativaZenchssug VaaotNessuna valutazione finora
- 2.informe Final de AmoniacoDocumento24 pagine2.informe Final de Amoniacobaneador5000% (1)
- DesarrollarDocumento151 pagineDesarrollarFelix Arias Hernandez100% (1)
- Instructivo Ficha Notificación SivimDocumento10 pagineInstructivo Ficha Notificación SivimLeonardo SuarezNessuna valutazione finora
- Sanación Cuantica SomosbuenavidaDocumento23 pagineSanación Cuantica SomosbuenavidaGrace SCNessuna valutazione finora
- Cartilla - PracticoNumero7 - Masas de Agua2021Documento9 pagineCartilla - PracticoNumero7 - Masas de Agua2021Guadalupe CascallaresNessuna valutazione finora
- Vivero Jaibana La Virginia 1233Documento13 pagineVivero Jaibana La Virginia 1233tillo1995Nessuna valutazione finora