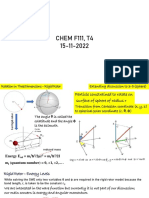
Potrebbero piacerti anche
- Guidelines For Winterization in Chemical PlantsDocumento11 pagineGuidelines For Winterization in Chemical Plantsmtrj59Nessuna valutazione finora
- 3.2 Air Conditioning of Buildings NotesDocumento7 pagine3.2 Air Conditioning of Buildings NotesDharshan K100% (1)
- Lecture 12Documento14 pagineLecture 12r prathapNessuna valutazione finora
- QMChap 8Documento9 pagineQMChap 8sanjeevchsNessuna valutazione finora
- Helium Atom, Approximate Methods: 24th April 2008Documento23 pagineHelium Atom, Approximate Methods: 24th April 2008Griffith AmakusaNessuna valutazione finora
- Vibrational SpectraDocumento27 pagineVibrational SpectraRodrigo Quintana ManfrediNessuna valutazione finora
- Lecture 8: Variational Method: The Helium Atom (10/13/2005) : A Few Comments On Chaotic SystemsDocumento8 pagineLecture 8: Variational Method: The Helium Atom (10/13/2005) : A Few Comments On Chaotic Systemsbgiangre8372Nessuna valutazione finora
- Helium Atom, Approximate Methods: 22nd April 2008Documento17 pagineHelium Atom, Approximate Methods: 22nd April 2008Julian David Henao EscobarNessuna valutazione finora
- 1 Lecture 2: The Helium AtomDocumento4 pagine1 Lecture 2: The Helium AtomGurvir SinghNessuna valutazione finora
- Hartree-Fock Theory: Erin Dahlke Department of Chemistry University of Minnesota Vlab Tutorial May 25, 2006Documento42 pagineHartree-Fock Theory: Erin Dahlke Department of Chemistry University of Minnesota Vlab Tutorial May 25, 2006Uttam PaliwalNessuna valutazione finora
- Quantum Mechanics, Chapter 8Documento7 pagineQuantum Mechanics, Chapter 8oneoonineNessuna valutazione finora
- Self-Consistent FieldDocumento6 pagineSelf-Consistent FieldmekokiNessuna valutazione finora
- Tight-Binding Model of Electronic StructuresDocumento9 pagineTight-Binding Model of Electronic StructuresJulian David Henao EscobarNessuna valutazione finora
- CHAPTER 10: Atomic Structure and Atomic SpectraDocumento25 pagineCHAPTER 10: Atomic Structure and Atomic SpectraVijay PradhanNessuna valutazione finora
- 08 TwoElectronDocumento78 pagine08 TwoElectronHamzaNessuna valutazione finora
- Bretonnet Rev BasicsDocumento34 pagineBretonnet Rev BasicsRahul RNessuna valutazione finora
- General Chemistry (CHEM F111) Lecture-11 13/04/2023Documento15 pagineGeneral Chemistry (CHEM F111) Lecture-11 13/04/2023Please Help BPHCNessuna valutazione finora
- Demtroeder Rotovibrazioni PDFDocumento23 pagineDemtroeder Rotovibrazioni PDFIrlani SismonikaNessuna valutazione finora
- Exam 1 & AnswersDocumento4 pagineExam 1 & AnswerspsychoxboyNessuna valutazione finora
- Pauli Exclusion PrincipleDocumento4 paginePauli Exclusion PrinciplePedroNessuna valutazione finora
- SelectionDocumento42 pagineSelectionAlejandra Ayulo CumpalliNessuna valutazione finora
- Identical Particles RevisitedDocumento6 pagineIdentical Particles Revisitedsam41206Nessuna valutazione finora
- C. David Sherrill - An Introduction To Hartree-Fock Molecular Orbital TheoryDocumento8 pagineC. David Sherrill - An Introduction To Hartree-Fock Molecular Orbital TheoryElectro_LiteNessuna valutazione finora
- Excitons in Bulk and Low-Dimensional SemiconductorsDocumento9 pagineExcitons in Bulk and Low-Dimensional Semiconductorsprakush_prakushNessuna valutazione finora
- Physical ChemistryDocumento190 paginePhysical ChemistryAdarsh PriyaranjanNessuna valutazione finora
- ch8 ProbsDocumento4 paginech8 ProbsEkrem GüldesteNessuna valutazione finora
- Time Dependent SEDocumento14 pagineTime Dependent SEabdullhNessuna valutazione finora
- Potential Scattering: 2. Also in This Section, We Consider The Threshold Behaviour WhenDocumento54 paginePotential Scattering: 2. Also in This Section, We Consider The Threshold Behaviour WhenAlessandra Souza BarbosaNessuna valutazione finora
- Exercises: Basic Quantum Mechanics: D DX D DXDocumento12 pagineExercises: Basic Quantum Mechanics: D DX D DXwsosornozNessuna valutazione finora
- Energy Band MethodsDocumento15 pagineEnergy Band MethodsharshitNessuna valutazione finora
- Hydrogen QMDocumento20 pagineHydrogen QMAmanpreetNessuna valutazione finora
- Approximation MethodsDocumento98 pagineApproximation Methodsbinseung skzNessuna valutazione finora
- Hitoshi QFTDocumento18 pagineHitoshi QFTgemunu271Nessuna valutazione finora
- Past Papers Solutions OutputDocumento35 paginePast Papers Solutions OutputCharlie Biopunk AlesNessuna valutazione finora
- Helium AtomDocumento8 pagineHelium AtomehmedNessuna valutazione finora
- BITS Pilani Chemistry Hamilton OperatorDocumento11 pagineBITS Pilani Chemistry Hamilton OperatorNaresh SehdevNessuna valutazione finora
- 2.01 The Schrodinger Wave Equation For The Hydrogen AtomDocumento10 pagine2.01 The Schrodinger Wave Equation For The Hydrogen AtomRumbaNessuna valutazione finora
- Identical Particles PDFDocumento31 pagineIdentical Particles PDFHồng NhânNessuna valutazione finora
- GCL 08Documento19 pagineGCL 08sahanishubham317Nessuna valutazione finora
- Atomic and Molecular Physics PH5410: Assignment 1Documento1 paginaAtomic and Molecular Physics PH5410: Assignment 1Suvashis MaityNessuna valutazione finora
- Solid State Physics NotesDocumento15 pagineSolid State Physics NotesSebastian YdeNessuna valutazione finora
- 01-The Integer Quantum Hall Effect I PDFDocumento7 pagine01-The Integer Quantum Hall Effect I PDFBheim LlonaNessuna valutazione finora
- Quantum Hall Effect - BDocumento7 pagineQuantum Hall Effect - Bspow123Nessuna valutazione finora
- MQ Cap7 PDFDocumento7 pagineMQ Cap7 PDFmekokiNessuna valutazione finora
- Diklic TNSDocumento17 pagineDiklic TNSJosipa DiklićNessuna valutazione finora
- Firouzifarrashbandi 2020Documento4 pagineFirouzifarrashbandi 2020kamal touilebNessuna valutazione finora
- Introduction To Computational Chemistry: Andrew S. IchimuraDocumento30 pagineIntroduction To Computational Chemistry: Andrew S. IchimuraAnonymous rn2qoBPjKyNessuna valutazione finora
- SCHR Odinger Equation For Central Potentials: 2.1 Variable SeparationDocumento9 pagineSCHR Odinger Equation For Central Potentials: 2.1 Variable SeparationJUAN SEBASTIÁN RAMÍREZ QUINTERONessuna valutazione finora
- Thanks To Yossef and Shiang Yong For Their Input in This ProblemDocumento8 pagineThanks To Yossef and Shiang Yong For Their Input in This ProblemIgnacio JuárezNessuna valutazione finora
- Fractional SCHR Odinger Wave Equation and Fractional Uncertainty PrincipleDocumento8 pagineFractional SCHR Odinger Wave Equation and Fractional Uncertainty PrincipleCristian CasaNessuna valutazione finora
- Rick Rajter: N N M K 2 I N NDocumento4 pagineRick Rajter: N N M K 2 I N Njaurav41Nessuna valutazione finora
- Hartree Fock TheoryDocumento8 pagineHartree Fock TheoryHoangVu3010Nessuna valutazione finora
- The Kosterlitz-Thouless TransitionDocumento14 pagineThe Kosterlitz-Thouless TransitionKenzaNessuna valutazione finora
- Exactly Model Particles Both Electric: Soluble Nonrelativistic of and Magnetic ChargesDocumento9 pagineExactly Model Particles Both Electric: Soluble Nonrelativistic of and Magnetic ChargesPhilippe NounahonNessuna valutazione finora
- Firewall or Smooth Horizon 1208Documento8 pagineFirewall or Smooth Horizon 1208Sk Ashif AkramNessuna valutazione finora
- Introduction To MagnetismDocumento29 pagineIntroduction To MagnetismJure MrduljasNessuna valutazione finora
- Chapter 08Documento12 pagineChapter 08Sabyasachi ChakrabortyNessuna valutazione finora
- 1 Symmetry of Potential and WavefunctionDocumento3 pagine1 Symmetry of Potential and WavefunctionHalloMannNessuna valutazione finora
- 97, Slow in Crystal: Electrons PolarDocumento6 pagine97, Slow in Crystal: Electrons PolarDejan DjokićNessuna valutazione finora
- Diatomic Molecule: Vibrational and Rotational SpectraDocumento16 pagineDiatomic Molecule: Vibrational and Rotational SpectraCamilo Monsalve MayaNessuna valutazione finora
- Effective Action and Renormalization Group Flow of Anisotropic SuperconductorsDocumento19 pagineEffective Action and Renormalization Group Flow of Anisotropic SuperconductorssatyabashaNessuna valutazione finora
- Advances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenDa EverandAdvances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenR. BrillNessuna valutazione finora
- Different Precipitation Response Over Land and Ocean To Orbital and Greenhouse Gas ForcingDocumento11 pagineDifferent Precipitation Response Over Land and Ocean To Orbital and Greenhouse Gas Forcingprivate3737Nessuna valutazione finora
- Zhang Et Al Measuring 2D MoS2 Refractive IndexDocumento7 pagineZhang Et Al Measuring 2D MoS2 Refractive Indexprivate3737Nessuna valutazione finora
- Photovoltaics PDFDocumento1 paginaPhotovoltaics PDFprivate3737Nessuna valutazione finora
- Photocurrent Micros PDFDocumento1 paginaPhotocurrent Micros PDFprivate3737Nessuna valutazione finora
- Adaptation of Gene Loci To Heterochromatin in The Course of Drosophila Evolution Is Associated With Insulator ProteinsDocumento18 pagineAdaptation of Gene Loci To Heterochromatin in The Course of Drosophila Evolution Is Associated With Insulator Proteinsprivate3737Nessuna valutazione finora
- Graphene Flagship 2018 ReportDocumento27 pagineGraphene Flagship 2018 Reportprivate3737Nessuna valutazione finora
- 2-Dimensional Semiconductors PDFDocumento1 pagina2-Dimensional Semiconductors PDFprivate3737Nessuna valutazione finora
- SSP Gnu FCC Lattice PDFDocumento1 paginaSSP Gnu FCC Lattice PDFprivate3737Nessuna valutazione finora
- This Is A File. Please Learn About The GNU Project. Solid State Physics Is Really InterestingDocumento1 paginaThis Is A File. Please Learn About The GNU Project. Solid State Physics Is Really Interestingprivate3737Nessuna valutazione finora
- This Is A File. Please Learn About The GNU ProjectDocumento1 paginaThis Is A File. Please Learn About The GNU Projectprivate3737Nessuna valutazione finora
- This Is A File. Please Learn About The GNU Project. Solid State Physics Is Really InterestingDocumento1 paginaThis Is A File. Please Learn About The GNU Project. Solid State Physics Is Really Interestingprivate3737Nessuna valutazione finora
- NanomatDocumento2 pagineNanomatprivate3737Nessuna valutazione finora
- This Is A PDF. Isn't That Great? Lorem IpsumDocumento1 paginaThis Is A PDF. Isn't That Great? Lorem Ipsumprivate3737Nessuna valutazione finora
- Information On LifeDocumento1 paginaInformation On Lifeprivate3737Nessuna valutazione finora
- PY251 SyllabusDocumento3 paginePY251 Syllabusprivate3737Nessuna valutazione finora
- This Is A PDFDocumento1 paginaThis Is A PDFprivate3737Nessuna valutazione finora
- InformationDocumento1 paginaInformationprivate3737Nessuna valutazione finora
- Spohr D MajorDocumento6 pagineSpohr D Majorprivate3737Nessuna valutazione finora
- Heat Transfer Detailed Lesson PlanDocumento20 pagineHeat Transfer Detailed Lesson PlanAiah Rica Sumalinog50% (2)
- 161 Lab Experiment 7 CalorimetryDocumento14 pagine161 Lab Experiment 7 CalorimetryInês LapaNessuna valutazione finora
- Organic ChemistryDocumento20 pagineOrganic ChemistryGirish RaguvirNessuna valutazione finora
- Monitoring Heat Exchanger Fouling For Optimal OperationDocumento4 pagineMonitoring Heat Exchanger Fouling For Optimal OperationlaythNessuna valutazione finora
- 11 Solutions 2aDocumento14 pagine11 Solutions 2amainethemaineNessuna valutazione finora
- Formula RioDocumento11 pagineFormula RioMoad BouzidaNessuna valutazione finora
- 7.2 Heat FlowDocumento50 pagine7.2 Heat FlowNITIASSWARENessuna valutazione finora
- Drying Problem SetDocumento1 paginaDrying Problem SetCaryl Faith GadianNessuna valutazione finora
- Material Balance Project Styrene Manufacture: H CHCH H C CH CH H CDocumento4 pagineMaterial Balance Project Styrene Manufacture: H CHCH H C CH CH H CMhd SakerNessuna valutazione finora
- Laboratory Manual in ES 15ADocumento2 pagineLaboratory Manual in ES 15AJay KayeNessuna valutazione finora
- Carbon NanotubesDocumento15 pagineCarbon NanotubesLaraib HabibNessuna valutazione finora
- Acidity, Alkalinity, and HardnessDocumento30 pagineAcidity, Alkalinity, and HardnessCharan DeepNessuna valutazione finora
- Question Bank - Part ADocumento5 pagineQuestion Bank - Part AvaishaliNessuna valutazione finora
- 12th Semester HL Physics TestDocumento20 pagine12th Semester HL Physics TestutheinsweNessuna valutazione finora
- FC Questions On Magnetic Materials and Its ClassificationsDocumento13 pagineFC Questions On Magnetic Materials and Its ClassificationsDawa PenjorNessuna valutazione finora
- Ice Plant Test RigDocumento9 pagineIce Plant Test Rigramniwas123Nessuna valutazione finora
- JEE Main Physics SyllabusDocumento1 paginaJEE Main Physics Syllabushari kishoreNessuna valutazione finora
- Flares CalculationsDocumento14 pagineFlares CalculationsAnonymous oVRvsdWzfBNessuna valutazione finora
- Combined Liquid Solutions FileDocumento357 pagineCombined Liquid Solutions FileMaloth ShruthiNessuna valutazione finora
- Heat Pipe in AHUDocumento2 pagineHeat Pipe in AHUjimmiilongNessuna valutazione finora
- Chapter 4 PhotochemistryDocumento19 pagineChapter 4 PhotochemistryHien HoangNessuna valutazione finora
- The Possiblillty of Negative KelvinDocumento6 pagineThe Possiblillty of Negative Kelvinapi-263357984Nessuna valutazione finora
- ATA 35 OxygenDocumento3 pagineATA 35 OxygenhmtuanbkNessuna valutazione finora
- Adsorption of Benzene and Toluene From Aqueous Solution Using A Composite Hydrogel of Alginate-Grafted With Mesoporous SilicaDocumento13 pagineAdsorption of Benzene and Toluene From Aqueous Solution Using A Composite Hydrogel of Alginate-Grafted With Mesoporous SilicaSITI NUR AFIQAH MAHAZANNessuna valutazione finora
- Dr. Sami El-Khatib American University of SharjahDocumento1 paginaDr. Sami El-Khatib American University of SharjahAmro HeshamNessuna valutazione finora
- DPP2 Coordination Compounds L-2Documento78 pagineDPP2 Coordination Compounds L-2IncNessuna valutazione finora
- Aatcc 81 (Actualizada-2022)Documento2 pagineAatcc 81 (Actualizada-2022)DIEGONessuna valutazione finora
- Chem 28 Fundamentals of Analytical ChemistryDocumento85 pagineChem 28 Fundamentals of Analytical ChemistryAcadGucciManeNessuna valutazione finora