Potrebbero piacerti anche
- 6 Fluid and Electrolytes Na, CL and K .6Documento6 pagine6 Fluid and Electrolytes Na, CL and K .6Raras MayangNessuna valutazione finora
- Absen Parenting ClassDocumento2 pagineAbsen Parenting ClassFadel BilondatuNessuna valutazione finora
- Sistematik Riviw ADHD PDFDocumento7 pagineSistematik Riviw ADHD PDFFadel BilondatuNessuna valutazione finora
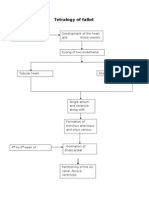
- Tetralogy Hypercyanotic SpellDocumento3 pagineTetralogy Hypercyanotic SpellJunior PratasikNessuna valutazione finora
- Chronic Hepatitis BDocumento13 pagineChronic Hepatitis BFadel BilondatuNessuna valutazione finora
- 443727Documento7 pagine443727Fadel BilondatuNessuna valutazione finora
- CopyrightDocumento2 pagineCopyrightmudasir61Nessuna valutazione finora
- Star BurnDocumento50 pagineStar BurnFadel BilondatuNessuna valutazione finora
- SaudiJKidneyDisTranspl27167-2006123 053421Documento6 pagineSaudiJKidneyDisTranspl27167-2006123 053421Fadel BilondatuNessuna valutazione finora
- Faktor Risiko Relaps Bandung 2016Documento3 pagineFaktor Risiko Relaps Bandung 2016Fadel BilondatuNessuna valutazione finora
- Tetralogy of FallotDocumento10 pagineTetralogy of Fallotwasiq_rawasiaNessuna valutazione finora
- Tetralogy of FallotDocumento10 pagineTetralogy of Fallotwasiq_rawasiaNessuna valutazione finora
- Overview of Hepatitis B Virus Infection in ChildrenDocumento14 pagineOverview of Hepatitis B Virus Infection in ChildrenFadel Bilondatu100% (1)
- Varzig Pi PDFDocumento12 pagineVarzig Pi PDFFadel BilondatuNessuna valutazione finora
- Overview of Hepatitis B Virus Infection in ChildrenDocumento14 pagineOverview of Hepatitis B Virus Infection in ChildrenFadel Bilondatu100% (1)
- 4-2-3-1 Wide ,, Gatling Project ,, The Invicible ,, 142 Goals in League - (Update)Documento8 pagine4-2-3-1 Wide ,, Gatling Project ,, The Invicible ,, 142 Goals in League - (Update)Fadel BilondatuNessuna valutazione finora
- Acid-Base Tutorial - Strong Ion DifferenceDocumento3 pagineAcid-Base Tutorial - Strong Ion DifferenceFadel BilondatuNessuna valutazione finora
- Protokol Soft Tissue SarcomaDocumento2 pagineProtokol Soft Tissue SarcomaFadel BilondatuNessuna valutazione finora
- TMH 0040 0362Documento7 pagineTMH 0040 0362Fadel BilondatuNessuna valutazione finora
- 19-Rapid Critical Appraisal of A Systematic ReviewDocumento2 pagine19-Rapid Critical Appraisal of A Systematic ReviewFadel BilondatuNessuna valutazione finora
- Jurnalku GiziDocumento6 pagineJurnalku GiziFadel BilondatuNessuna valutazione finora
- 14 Artikel MetaDocumento6 pagine14 Artikel MetaFadel BilondatuNessuna valutazione finora
- Liquidos y Electrolitos 2Documento4 pagineLiquidos y Electrolitos 2Jorge MBNessuna valutazione finora
- Transfusion Support in Patients With Dengue FeverDocumento7 pagineTransfusion Support in Patients With Dengue FeverFadel BilondatuNessuna valutazione finora
- Jurnalku GiziDocumento6 pagineJurnalku GiziFadel BilondatuNessuna valutazione finora
- Efficacy of Platelet Transfusions in Immune ThrombocytopeniaDocumento4 pagineEfficacy of Platelet Transfusions in Immune ThrombocytopeniaFadel BilondatuNessuna valutazione finora
- 100008IJBTINK2012 KulkarniDocumento5 pagine100008IJBTINK2012 KulkarniFadel BilondatuNessuna valutazione finora
- 10 1146@annurev Me 31 020180 002453Documento32 pagine10 1146@annurev Me 31 020180 002453Fadel BilondatuNessuna valutazione finora
- 862 FullDocumento3 pagine862 FullFadel BilondatuNessuna valutazione finora
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5784)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (72)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (119)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Tetralogy of FallotDocumento3 pagineTetralogy of FallotJohn Mark PocsidioNessuna valutazione finora
- Tetralogy of FallotDocumento7 pagineTetralogy of FallotAshok Kumar JangirNessuna valutazione finora
- MT 1Documento57 pagineMT 1Bibek goitNessuna valutazione finora
- Maternal and Child Health Nursing 4Documento106 pagineMaternal and Child Health Nursing 4Vin Santos33% (3)
- Pathophysiology and Treatment of Tetralogy of FallotDocumento2 paginePathophysiology and Treatment of Tetralogy of FallotMark James Melendres100% (12)
- Cyanotic Congenital Heart DiseaseDocumento22 pagineCyanotic Congenital Heart DiseaseRaviNessuna valutazione finora
- Conginital Heart DiseaseDocumento19 pagineConginital Heart DiseaseSanthosh.S.UNessuna valutazione finora
- Plab 1 NotesDocumento141 paginePlab 1 Notesthiri sakura100% (3)
- Christopher Cheung Approach To Cardiac History Taking: Cyanosis or SyncopeDocumento6 pagineChristopher Cheung Approach To Cardiac History Taking: Cyanosis or SyncopeApryl Phyllis JimenezNessuna valutazione finora
- PEDIATRIC NURSING ASSESSMENT EXAM MAY 2023Documento12 paginePEDIATRIC NURSING ASSESSMENT EXAM MAY 2023AnnJelicaAbonNessuna valutazione finora
- Tetralogy of FallotDocumento22 pagineTetralogy of FallotHusna Aje100% (1)
- DR - Azad A Haleem AL - Brefkani: University of Duhok Faculty of Medical Science School of Medicine Pediatrics DepartmentDocumento58 pagineDR - Azad A Haleem AL - Brefkani: University of Duhok Faculty of Medical Science School of Medicine Pediatrics DepartmentGomathi ShankarNessuna valutazione finora
- Early Brain Changes in Ischemic StrokeDocumento25 pagineEarly Brain Changes in Ischemic StrokeVerónica BezaresNessuna valutazione finora
- Pathoma Lecture Notes 2017Documento42 paginePathoma Lecture Notes 2017Priyesh PrinceNessuna valutazione finora
- Pediatric Echocardiography TechniquesDocumento17 paginePediatric Echocardiography Techniquesraviks34Nessuna valutazione finora
- Pedia AbDocumento19 paginePedia AbYaj CruzadaNessuna valutazione finora
- Embryology QuestionDocumento19 pagineEmbryology QuestionVarun Arunagiri100% (17)
- Case60 Down SyndromeDocumento9 pagineCase60 Down Syndromegreenflames09100% (2)
- Congenital Heart Surgery ManagementDocumento9 pagineCongenital Heart Surgery ManagementprofarmahNessuna valutazione finora
- Differences in Morbidity and Mortality in Down Syndrome Are Related To The Type of Congenital Heart DefectDocumento10 pagineDifferences in Morbidity and Mortality in Down Syndrome Are Related To The Type of Congenital Heart DefectMaría BonettiNessuna valutazione finora
- Scans en 2015Documento43 pagineScans en 2015Carolina Duque RodriguezNessuna valutazione finora
- Tetralogy of FallotDocumento5 pagineTetralogy of FallotNoor FadhilaNessuna valutazione finora
- Congenital Heart DiseasesDocumento40 pagineCongenital Heart DiseasesiooaNessuna valutazione finora
- Pediatric Quick NotesDocumento178 paginePediatric Quick NotesCharlotte McDonald100% (5)
- Cyanotic Congenital Heart DiseaseDocumento6 pagineCyanotic Congenital Heart DiseaseSimran JosanNessuna valutazione finora
- 10 Common EKG Heart RhythmsDocumento204 pagine10 Common EKG Heart RhythmsJames BrooksherNessuna valutazione finora
- Cyanotic Spells (Tet Spells) PDFDocumento18 pagineCyanotic Spells (Tet Spells) PDFIndri Pratiwi TobingNessuna valutazione finora
- Care of High Risk Newborn - ChaboyDocumento9 pagineCare of High Risk Newborn - Chaboychfalguera0% (1)
- PreviewpdfDocumento235 paginePreviewpdfchubura_je_rajNessuna valutazione finora