Potrebbero piacerti anche
- Legal Collaboration Agreement OverviewDocumento6 pagineLegal Collaboration Agreement OverviewAkash GoelNessuna valutazione finora
- MasqueradeDocumento1 paginaMasqueradeAkash GoelNessuna valutazione finora
- Certificate: Ayurvedic & Unani Tibbia CollegeDocumento4 pagineCertificate: Ayurvedic & Unani Tibbia CollegeAkash GoelNessuna valutazione finora
- Aiats 5-Unlocked PDFDocumento40 pagineAiats 5-Unlocked PDFAkash Goel100% (1)
- Chapter36 - BiomoleculesDocumento19 pagineChapter36 - BiomoleculesAkash GoelNessuna valutazione finora
- 0901 - Physics - Paper-With Ans-Solution - MorningDocumento9 pagine0901 - Physics - Paper-With Ans-Solution - MorningAkash GoelNessuna valutazione finora
- 0901 - Chemistry - Paper-With Ans-Solution - EveningDocumento8 pagine0901 - Chemistry - Paper-With Ans-Solution - EveningAkash GoelNessuna valutazione finora
- Chapter 13Documento52 pagineChapter 13Muhammad UsmanNessuna valutazione finora
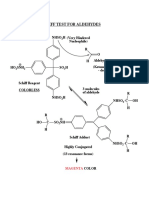
- Detect aldehydes with Schiff reagent testDocumento1 paginaDetect aldehydes with Schiff reagent testAkash GoelNessuna valutazione finora
- Module2 Reduction PDFDocumento55 pagineModule2 Reduction PDFAnonymous vRpzQ2BLNessuna valutazione finora
- August Biology TodayDocumento22 pagineAugust Biology TodayAkash GoelNessuna valutazione finora
- CamScanner Document ScansDocumento47 pagineCamScanner Document ScansAkash GoelNessuna valutazione finora
- JEE Main Advanced P I PHYSICS Paper Answer PDFDocumento6 pagineJEE Main Advanced P I PHYSICS Paper Answer PDFAkash GoelNessuna valutazione finora
- Admission List - Mopup Round PG CounsellingDocumento3 pagineAdmission List - Mopup Round PG CounsellingAkash GoelNessuna valutazione finora
- Chapter36 - Biomolecules PDFDocumento34 pagineChapter36 - Biomolecules PDFAkash GoelNessuna valutazione finora
- Surety Bond 2017Documento3 pagineSurety Bond 2017Akash GoelNessuna valutazione finora
- B.sc. Hons. ZoologyDocumento110 pagineB.sc. Hons. ZoologyHarsh Dalal0% (1)
- PDFDocumento249 paginePDFAkash GoelNessuna valutazione finora
- KMC MPL AttachementDocumento8 pagineKMC MPL AttachementAkash GoelNessuna valutazione finora
- Class Test SolutionDocumento10 pagineClass Test SolutionAkash GoelNessuna valutazione finora
- AP Eamcet 2015Documento43 pagineAP Eamcet 2015Akash GoelNessuna valutazione finora
- HW 10 AkeyDocumento9 pagineHW 10 AkeyAkash GoelNessuna valutazione finora
- Plancess Study Material Mathematics For Jee Set 9788126555758 PDFDocumento3 paginePlancess Study Material Mathematics For Jee Set 9788126555758 PDFAkash GoelNessuna valutazione finora
- Derivation of Clausius Clapeyron Equation PDFDocumento1 paginaDerivation of Clausius Clapeyron Equation PDFShweta SridharNessuna valutazione finora
- Mbbs 2017 ListDocumento7 pagineMbbs 2017 ListAkash GoelNessuna valutazione finora
- Schedule For Admission To Mbbs Course at Afmc, Pune For Year 2017Documento1 paginaSchedule For Admission To Mbbs Course at Afmc, Pune For Year 2017Akash GoelNessuna valutazione finora
- E-Prospectus 2018-19 PDFDocumento225 pagineE-Prospectus 2018-19 PDFKishan VarshneyNessuna valutazione finora
- Final Ug 2017 Counseling Schedule FinalDocumento1 paginaFinal Ug 2017 Counseling Schedule FinalAkash GoelNessuna valutazione finora
- (WWW - Entrance Exam - Net) IcarDocumento5 pagine(WWW - Entrance Exam - Net) IcarAkash GoelNessuna valutazione finora
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5783)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (890)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (265)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (72)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (119)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- 5054 s16 QP 42 PDFDocumento12 pagine5054 s16 QP 42 PDFInjamam Ul HaqueNessuna valutazione finora
- Newton's Laws of Motion ExperimentDocumento3 pagineNewton's Laws of Motion ExperimentMartie DuranaNessuna valutazione finora
- Force and acceleration problem setDocumento2 pagineForce and acceleration problem setBenedick Jayson P. Marti100% (1)
- Index of Physics Lab Report on Refraction of LightDocumento6 pagineIndex of Physics Lab Report on Refraction of LightSnehal VinodNessuna valutazione finora
- Synthesis of AspirinDocumento3 pagineSynthesis of AspirinShoomyla RashidNessuna valutazione finora
- Hecke's L-FunctionsDocumento102 pagineHecke's L-FunctionsluisufspaiandreNessuna valutazione finora
- Fossen - Chapter 3: Strain in RocksDocumento41 pagineFossen - Chapter 3: Strain in RocksbuhlejoyNessuna valutazione finora
- "Explain With Example That Rate of Convergence of False Position Method Is Faster Than That of The Bisection Method.Documento3 pagine"Explain With Example That Rate of Convergence of False Position Method Is Faster Than That of The Bisection Method.gauravsanadhyaNessuna valutazione finora
- Phys 1004 - Cheat - Sheet. Electromagnetism and WavesDocumento2 paginePhys 1004 - Cheat - Sheet. Electromagnetism and WavesNadya B.Nessuna valutazione finora
- 293 Tanuja Ipwh Assignment 1Documento2 pagine293 Tanuja Ipwh Assignment 1Tanuja MasginNessuna valutazione finora
- ISO 8573 Purity Classes PDFDocumento1 paginaISO 8573 Purity Classes PDFOky Andytya PratamaNessuna valutazione finora
- DS-500 Series Weighing ScaleDocumento1 paginaDS-500 Series Weighing ScaleAntmavrNessuna valutazione finora
- Applus EN 13501-1 Test Report Part 1 PDFDocumento24 pagineApplus EN 13501-1 Test Report Part 1 PDFaziz hNessuna valutazione finora
- OMAE2018 ProgramDocumento124 pagineOMAE2018 ProgramTahsin TezdoganNessuna valutazione finora
- Numerical Method 1Documento3 pagineNumerical Method 1Er SarbeshNessuna valutazione finora
- Theoretical Physics 1: Brwebberandchwbarnes Michaelmas Term 2008Documento73 pagineTheoretical Physics 1: Brwebberandchwbarnes Michaelmas Term 200821260paco61Nessuna valutazione finora
- Relevance of Research in Social WorkDocumento15 pagineRelevance of Research in Social WorkangelgijoNessuna valutazione finora
- Electron Configuration NotesDocumento4 pagineElectron Configuration NotesapriantokaNessuna valutazione finora
- Soil Bearing CapacityDocumento33 pagineSoil Bearing CapacityAce JokerNessuna valutazione finora
- Doob, The Development of Rigor in Mathematical Probability (1900-1950) PDFDocumento11 pagineDoob, The Development of Rigor in Mathematical Probability (1900-1950) PDFFardadPouranNessuna valutazione finora
- 4439testing MultipleDocumento4 pagine4439testing MultipleSonny RamosNessuna valutazione finora
- Chapter 4Documento9 pagineChapter 4dearsaswatNessuna valutazione finora
- OuzineDocumento4 pagineOuzineNima ArvinNessuna valutazione finora
- Precast Bridge DeckDocumento4 paginePrecast Bridge DeckPaul_delgadoNessuna valutazione finora
- Analytical Techniques Julia C. Drees Alan H. B. WuDocumento36 pagineAnalytical Techniques Julia C. Drees Alan H. B. WuBetrearon SileshiNessuna valutazione finora
- Is - 807Documento47 pagineIs - 807Mohit Arora0% (1)
- Bonfire Certificate: Priyabrat Prasd Mohapatra Roll No.Documento22 pagineBonfire Certificate: Priyabrat Prasd Mohapatra Roll No.Priyabrat PrasadNessuna valutazione finora
- Inroduction To AnysisDocumento7 pagineInroduction To Anysissantosh gorliNessuna valutazione finora
- MA STEM Grade 8 Practice Test GuideDocumento30 pagineMA STEM Grade 8 Practice Test GuideRG Emmanuel TesaNessuna valutazione finora
- Three Major Design PhilosophiesDocumento4 pagineThree Major Design PhilosophiesFatima Ahmed100% (1)