Potrebbero piacerti anche
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- Cell Culture Basics HandbookDocumento120 pagineCell Culture Basics HandbookbrunafariassNessuna valutazione finora
- Handbook For Cell Culture Basics (Gibco)Documento62 pagineHandbook For Cell Culture Basics (Gibco)Cesar PuentesNessuna valutazione finora
- Sag Slurry PoolingDocumento10 pagineSag Slurry PoolingalgroneNessuna valutazione finora
- Avr GeneralDocumento67 pagineAvr GeneralRukma Goud Shakkari100% (2)
- Revised Design Report of Jetty 06.04.2014Documento10 pagineRevised Design Report of Jetty 06.04.2014Priodeep Chowdhury100% (2)
- Latent Print DevelopmentDocumento20 pagineLatent Print Developmentapi-272036460100% (1)
- GE Gas Turbine IGV AngleDocumento10 pagineGE Gas Turbine IGV AngleSamir BenabdallahNessuna valutazione finora
- Waste Incineration Insert v2Documento2 pagineWaste Incineration Insert v2Namik HadziibrahimovicNessuna valutazione finora
- Cute Exam Paper Set 4Documento12 pagineCute Exam Paper Set 4Budak Baru BelajarNessuna valutazione finora
- Set 2 ENGLISH Question SolvedDocumento19 pagineSet 2 ENGLISH Question SolvedBudak Baru BelajarNessuna valutazione finora
- Chartered Scientist (CSci)Documento1 paginaChartered Scientist (CSci)Budak Baru BelajarNessuna valutazione finora
- Webinar scriptsDocumento2 pagineWebinar scriptsNurul FadilaNessuna valutazione finora
- IndexDocumento13 pagineIndexBudak Baru BelajarNessuna valutazione finora
- Comparison of Most Probable Number-PCR and Most Probable Number-Foci Detection Method For Quantifying Infectious Cryptosporidium Parvum Oocysts in Environmental SamplesDocumento10 pagineComparison of Most Probable Number-PCR and Most Probable Number-Foci Detection Method For Quantifying Infectious Cryptosporidium Parvum Oocysts in Environmental SamplesBudak Baru BelajarNessuna valutazione finora
- Sekolah Kurang MuridDocumento11 pagineSekolah Kurang MuridBudak Baru BelajarNessuna valutazione finora
- Contributors from Heriot-Watt University, Ryerson University, Scottish Water and moreDocumento1 paginaContributors from Heriot-Watt University, Ryerson University, Scottish Water and moreBudak Baru BelajarNessuna valutazione finora
- Salasilah Raja Reman5Documento1 paginaSalasilah Raja Reman5Budak Baru BelajarNessuna valutazione finora
- ARTIFICIAL DOUBLE PNEUMOTHORAX CASE STUDYDocumento2 pagineARTIFICIAL DOUBLE PNEUMOTHORAX CASE STUDYBudak Baru BelajarNessuna valutazione finora
- Waterborne Pathogens: Detection Methods and ApplicationsDocumento1 paginaWaterborne Pathogens: Detection Methods and ApplicationsBudak Baru BelajarNessuna valutazione finora
- Comparison of Efficiency of Various DNA Extraction Methods From Cysts of Giardia Intestinalis Measured by PCR and TaqMan Real Time PCRDocumento8 pagineComparison of Efficiency of Various DNA Extraction Methods From Cysts of Giardia Intestinalis Measured by PCR and TaqMan Real Time PCRBudak Baru BelajarNessuna valutazione finora
- AcknowledgmentDocumento1 paginaAcknowledgmentBudak Baru BelajarNessuna valutazione finora
- 5 Reasons Cancer Researchers Adopt 3D Cell Culture White PaperDocumento19 pagine5 Reasons Cancer Researchers Adopt 3D Cell Culture White PaperBudak Baru BelajarNessuna valutazione finora
- Introduction to Investment-Linked InsuranceDocumento197 pagineIntroduction to Investment-Linked InsuranceSitisaiful0% (1)
- Validation of Cell-Free Culture Using Scanning Electron Microscopy (SEM) and Gene Expression StudiesDocumento9 pagineValidation of Cell-Free Culture Using Scanning Electron Microscopy (SEM) and Gene Expression StudiesBudak Baru BelajarNessuna valutazione finora
- Comprehensive Guide to Unit Trust FundamentalsDocumento132 pagineComprehensive Guide to Unit Trust FundamentalsMohd Hatif KamailNessuna valutazione finora
- Recent Advances in The Developmental Biology and Life Cycle of CryptosporidiumDocumento13 pagineRecent Advances in The Developmental Biology and Life Cycle of CryptosporidiumBudak Baru BelajarNessuna valutazione finora
- Cute Exam Paper Set 5Documento12 pagineCute Exam Paper Set 5Budak Baru BelajarNessuna valutazione finora
- Cute Exam Paper Set 1Documento20 pagineCute Exam Paper Set 1Budak Baru Belajar0% (1)
- Cute Exam Paper Set 3Documento18 pagineCute Exam Paper Set 3Budak Baru Belajar100% (1)
- Karkun 8Documento1 paginaKarkun 8Budak Baru BelajarNessuna valutazione finora
- Karkun 9Documento1 paginaKarkun 9Budak Baru BelajarNessuna valutazione finora
- Medicinal OrchidsDocumento14 pagineMedicinal OrchidsBudak Baru BelajarNessuna valutazione finora
- Cute Exam Paper Set 1Documento20 pagineCute Exam Paper Set 1Budak Baru Belajar0% (1)
- Kitab Turath1Documento11 pagineKitab Turath1Budak Baru Belajar100% (1)
- 2013/14 Academic Calendar Year 1 Mbbs 220 Programme, Faculty of Medicine UitmDocumento1 pagina2013/14 Academic Calendar Year 1 Mbbs 220 Programme, Faculty of Medicine UitmBudak Baru BelajarNessuna valutazione finora
- McCabe-Thiele Diagrams For Binary DistillationDocumento8 pagineMcCabe-Thiele Diagrams For Binary DistillationwetcoNessuna valutazione finora
- ME 555 Stress Analysis Unit 4Documento57 pagineME 555 Stress Analysis Unit 4TheoNessuna valutazione finora
- EPA 1668 A, Ag-2003Documento129 pagineEPA 1668 A, Ag-2003Karina Rondon RivadeneyraNessuna valutazione finora
- DPP-1 QuantizationDocumento1 paginaDPP-1 QuantizationVikasNessuna valutazione finora
- General Physics 1 1st Quarter Module 1 ActivitiesDocumento16 pagineGeneral Physics 1 1st Quarter Module 1 ActivitiesMica LopezNessuna valutazione finora
- Hydrostatic Forces on SurfacesDocumento12 pagineHydrostatic Forces on SurfacesPajhmanAwghanNessuna valutazione finora
- Abdel Jawad 2005Documento8 pagineAbdel Jawad 2005Alberto Tupa OrtizNessuna valutazione finora
- Fourier Series ApplicationDocumento10 pagineFourier Series Application9th P/C completedNessuna valutazione finora
- The Mode of Eruptions and Their Tephra Deposits: Tetsuo K and Mitsuru ODocumento8 pagineThe Mode of Eruptions and Their Tephra Deposits: Tetsuo K and Mitsuru OAnggit Tri AtmajaNessuna valutazione finora
- Folder Airless Auto Serie AlDocumento2 pagineFolder Airless Auto Serie AlErika MaraNessuna valutazione finora
- Precision Thermometers CatalogueDocumento44 paginePrecision Thermometers CataloguemarthaNessuna valutazione finora
- Lightning Protection Systems Advantages and DisadvantagesDocumento11 pagineLightning Protection Systems Advantages and DisadvantagesRamiro Magbanua FelicianoNessuna valutazione finora
- Key Words: Targeting, HEN, Composite Curve,: Module 04: Targeting Lecture 10: Energy Targeting ProcedureDocumento8 pagineKey Words: Targeting, HEN, Composite Curve,: Module 04: Targeting Lecture 10: Energy Targeting ProcedureCalNessuna valutazione finora
- Curriculum-Of Mathematics Government College Women University, SialkotDocumento119 pagineCurriculum-Of Mathematics Government College Women University, SialkotHuzaifa GurmaniNessuna valutazione finora
- Unit Hydrograph DerivationDocumento7 pagineUnit Hydrograph DerivationSudharsananPRSNessuna valutazione finora
- MFIX On of Discrete Element MethodDocumento30 pagineMFIX On of Discrete Element MethodkamranianNessuna valutazione finora
- EagleBurgmann Statotherm P Foil 9591 P enDocumento1 paginaEagleBurgmann Statotherm P Foil 9591 P enkeyur1109Nessuna valutazione finora
- Adrian Stan MFQMCourseHsL2006Documento60 pagineAdrian Stan MFQMCourseHsL2006禿公Nessuna valutazione finora
- Mohit SIR LATEST Notes (GATE+ESE-2020) )Documento5 pagineMohit SIR LATEST Notes (GATE+ESE-2020) )Vipul MetaNessuna valutazione finora
- About The Company: Machined and Forged ComponentsDocumento18 pagineAbout The Company: Machined and Forged ComponentsankitNessuna valutazione finora
- LaminateDocumento154 pagineLaminateAbhishek VaggarNessuna valutazione finora
- Schrodinger Equation DerivationDocumento12 pagineSchrodinger Equation DerivationAndrés López Martínez100% (1)
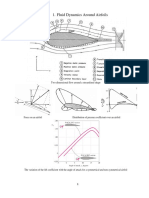
- Overview Aerodynamics 2017Documento10 pagineOverview Aerodynamics 2017marcoNessuna valutazione finora
- Kinematics of Machinery: Motion and AnalysisDocumento29 pagineKinematics of Machinery: Motion and AnalysisShashank SinghNessuna valutazione finora