Potrebbero piacerti anche
- Human Metabolism MapDocumento1 paginaHuman Metabolism MapPkern83% (6)
- Policy MemoDocumento4 paginePolicy Memoapi-340053591Nessuna valutazione finora
- Equations For Primary FRCADocumento11 pagineEquations For Primary FRCAMustafa SharkawyNessuna valutazione finora
- Equations For Primary FRCADocumento11 pagineEquations For Primary FRCAMustafa SharkawyNessuna valutazione finora
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocumento10 pagineThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernNessuna valutazione finora
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocumento10 pagineThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernNessuna valutazione finora
- The Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFDocumento10 pagineThe Crashing Ventilated Patient Ch.3 (Jairo I. Santanilla, ACEP, 2011) PDFPkernNessuna valutazione finora
- NUR 3032 Immune System Study PlanDocumento5 pagineNUR 3032 Immune System Study PlanThalia Fortune100% (1)
- The Crashing Ventilated Patient Algorthm (Jairo I. Santanilla, ACEP, 2011)Documento1 paginaThe Crashing Ventilated Patient Algorthm (Jairo I. Santanilla, ACEP, 2011)PkernNessuna valutazione finora
- 6th Central Pay Commission Salary CalculatorDocumento15 pagine6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Erotic CountertransferenceDocumento3 pagineErotic CountertransferenceVictor Gan100% (2)
- Gabbard, Transference and CountertransferenceDocumento9 pagineGabbard, Transference and Countertransferencejuaromer100% (1)
- Cholinergic DrugsDocumento15 pagineCholinergic DrugsChris Girgis100% (1)
- Respiratory-Equations (Adam Hollingworth)Documento4 pagineRespiratory-Equations (Adam Hollingworth)PkernNessuna valutazione finora
- Energy Drink AssignmentDocumento4 pagineEnergy Drink AssignmentSukh Prabhleen DhaliwalNessuna valutazione finora
- Tka Inservice HandoutDocumento2 pagineTka Inservice Handoutapi-317395769Nessuna valutazione finora
- KINESIOTHERAPHYDocumento40 pagineKINESIOTHERAPHYJeyarajasekar TtrNessuna valutazione finora
- Nurses NotesDocumento1 paginaNurses Notesmona_javier100% (4)
- Lower Limb FracturesDocumento124 pagineLower Limb FracturesMaríaJosé Dip100% (3)
- Chapter 4 Pharmacotherapeutics, Dynamics & KineticsDocumento47 pagineChapter 4 Pharmacotherapeutics, Dynamics & KineticsAshaNessuna valutazione finora
- Adrenergic AgonistsDocumento18 pagineAdrenergic AgonistsJod Bell100% (1)
- Pharmacology Assignment No.02: Submitted By: Submitted To: Nandraj Ma'am Areeba Shafiq Roll No. 1817007Documento23 paginePharmacology Assignment No.02: Submitted By: Submitted To: Nandraj Ma'am Areeba Shafiq Roll No. 1817007Nandraj123100% (1)
- Assessing Neurologic SystemDocumento7 pagineAssessing Neurologic Systemjanikkakristal100% (1)
- The Leukotrienes: Chemistry and BiologyDa EverandThe Leukotrienes: Chemistry and BiologyLawrence ChakrinNessuna valutazione finora
- Lipid Lowering DrugsDocumento15 pagineLipid Lowering Drugsmwaithira71682Nessuna valutazione finora
- Pharmaco DynamicsDocumento7 paginePharmaco DynamicsDavid NicholasNessuna valutazione finora
- Pharmacokinetic Study Guide LehneDocumento14 paginePharmacokinetic Study Guide LehneEllen HenningsNessuna valutazione finora
- Site of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFDocumento1 paginaSite of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFSunilNessuna valutazione finora
- Physiology of CoagulationDocumento44 paginePhysiology of CoagulationXee JayNessuna valutazione finora
- DIC Student Case StudyDocumento4 pagineDIC Student Case StudyJenn GallowayNessuna valutazione finora
- Clotting Concept Analysis Diagram and ExplanationDocumento2 pagineClotting Concept Analysis Diagram and ExplanationJulius Haynes100% (1)
- Antidepressants: Depression Is One The Most Treatable Mental IllnessDocumento40 pagineAntidepressants: Depression Is One The Most Treatable Mental IllnessMohammed AbdullahNessuna valutazione finora
- Adrenal Insufficiency and Cushing's Disease-1Documento34 pagineAdrenal Insufficiency and Cushing's Disease-1Mwanja MosesNessuna valutazione finora
- Cytologic Patterns - Eclinpath PDFDocumento5 pagineCytologic Patterns - Eclinpath PDFJD46Nessuna valutazione finora
- Antineoplastic DrugsDocumento16 pagineAntineoplastic DrugstheintrovNessuna valutazione finora
- Anti Depressant Drug LatestDocumento65 pagineAnti Depressant Drug LatestAnonymous zOMkw9100% (1)
- Chapter 73: Drug Therapy of Rheumatoid Arthritis Test Bank: Multiple ChoiceDocumento4 pagineChapter 73: Drug Therapy of Rheumatoid Arthritis Test Bank: Multiple ChoiceNurse Utopia100% (1)
- GPCRDocumento32 pagineGPCRSergio UribeNessuna valutazione finora
- 15G Protein-Coupled ReceptorDocumento15 pagine15G Protein-Coupled ReceptorZiedTrikiNessuna valutazione finora
- Osteomyelitis 130708212636 Phpapp01Documento107 pagineOsteomyelitis 130708212636 Phpapp01merikasorNessuna valutazione finora
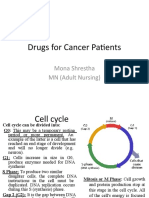
- Drugs For Cancer Patients: Mona Shrestha MN (Adult Nursing)Documento77 pagineDrugs For Cancer Patients: Mona Shrestha MN (Adult Nursing)sushma shresthaNessuna valutazione finora
- AnaemiaDocumento40 pagineAnaemiaNaveen Kumar100% (1)
- Pharmacokinetics PDFDocumento14 paginePharmacokinetics PDFLea PesiganNessuna valutazione finora
- GoutDocumento4 pagineGoutapi-3822433100% (1)
- TBI FinalDocumento28 pagineTBI Finalawais mpNessuna valutazione finora
- Pharmacology Module PDFDocumento23 paginePharmacology Module PDFmirza_baig_46100% (1)
- Adrenergic AgonistsDocumento43 pagineAdrenergic Agonistsmatchees-gone rogueNessuna valutazione finora
- LocalanestheticsDocumento51 pagineLocalanestheticskingkb4uNessuna valutazione finora
- 1introDocumento158 pagine1introDea MaharanisNessuna valutazione finora
- Drugs Used in TuberculosisDocumento27 pagineDrugs Used in Tuberculosisapi-3705123Nessuna valutazione finora
- Pharmacology Pharmacokinetics and Pharmacodynamics - PPT - Dr. Maulana Antian Empitu (Airlangga Medical Faculty)Documento59 paginePharmacology Pharmacokinetics and Pharmacodynamics - PPT - Dr. Maulana Antian Empitu (Airlangga Medical Faculty)rizkyyunitaa15Nessuna valutazione finora
- ANS ReceptorsDocumento14 pagineANS ReceptorsAisha AliNessuna valutazione finora
- Antiarrhythmic Drugs Classification (Vaughan Williams)Documento8 pagineAntiarrhythmic Drugs Classification (Vaughan Williams)ana100% (1)
- AlcoholsDocumento23 pagineAlcoholsdhaineyNessuna valutazione finora
- Adverse Drug Reactions (ADRS)Documento24 pagineAdverse Drug Reactions (ADRS)ANAM IQBALNessuna valutazione finora
- Sepsis and Septic Shock: Elise Mittleman Boller, - Cynthia M. OttoDocumento9 pagineSepsis and Septic Shock: Elise Mittleman Boller, - Cynthia M. OttoIan SabogalNessuna valutazione finora
- Top 5 Leukogram PatternsDocumento3 pagineTop 5 Leukogram PatternsSandraHermosilloNessuna valutazione finora
- Cholinergic DrugsDocumento44 pagineCholinergic Drugskhuzaima9100% (1)
- Clinical Haematology-Lecture SlidesDocumento55 pagineClinical Haematology-Lecture SlidesShiv Sookun100% (1)
- Mayra Pagan: Pharmacology-NursingDocumento50 pagineMayra Pagan: Pharmacology-NursingmayraNessuna valutazione finora
- Uptake and Distribution of Volatile AnestheticsDocumento22 pagineUptake and Distribution of Volatile AnestheticsSuresh Kumar100% (3)
- Asthma: A. Practice EssentialsDocumento8 pagineAsthma: A. Practice EssentialsCandha NurcahyaNessuna valutazione finora
- Disorder of The Neuromuscular Junction: Courtesy . DR - Syeda Afsheen Hasnain DPT/MSPT NeuroloicalDocumento16 pagineDisorder of The Neuromuscular Junction: Courtesy . DR - Syeda Afsheen Hasnain DPT/MSPT NeuroloicalCHANGEZ KHAN SARDAR100% (1)
- Fluid and ElyctrolyteDocumento49 pagineFluid and Elyctrolyteuuuhbnb lplhghNessuna valutazione finora
- Nervous System: Chapter # 7Documento69 pagineNervous System: Chapter # 7saddam ud dinNessuna valutazione finora
- 3 NeurotransmissionDocumento31 pagine3 Neurotransmissionsarahcho12152Nessuna valutazione finora
- Medication - ALT-Template - Enoxaparin SodiumDocumento1 paginaMedication - ALT-Template - Enoxaparin SodiumNancyAmissahNessuna valutazione finora
- Trypanosoma: Sri SundariDocumento37 pagineTrypanosoma: Sri SundariVaniaNessuna valutazione finora
- Pharmacological TermsDocumento18 paginePharmacological TermsMpdoNessuna valutazione finora
- Innate ImmunityDocumento9 pagineInnate Immunitynascha dumpNessuna valutazione finora
- Tissue TransplantDocumento20 pagineTissue TransplantEva Boje-JugadorNessuna valutazione finora
- Syncope: - Selvarathi KDocumento27 pagineSyncope: - Selvarathi KSelvarathi KandhaswamyNessuna valutazione finora
- CNS DrugsDocumento8 pagineCNS DrugsSheral Aida100% (2)
- Carbohydrate and Lipid MetabolismDocumento47 pagineCarbohydrate and Lipid MetabolismShirley Faye SalesNessuna valutazione finora
- Myasthenia GravisDocumento33 pagineMyasthenia GravisLovely Cervantes100% (1)
- Lecture 1 PDFDocumento75 pagineLecture 1 PDFBasil Elbushra Ahmed DomiNessuna valutazione finora
- Atlas of Small Animal Wound Management and Reconstructive SurgeryDa EverandAtlas of Small Animal Wound Management and Reconstructive SurgeryNessuna valutazione finora
- Simple Adrenal Synthesis PathwayDocumento1 paginaSimple Adrenal Synthesis PathwayPkernNessuna valutazione finora
- Selecting The Right' Positive End-Expiratory Pressure Level (Luciano Gattinoni, Current Opinion, 2016)Documento8 pagineSelecting The Right' Positive End-Expiratory Pressure Level (Luciano Gattinoni, Current Opinion, 2016)PkernNessuna valutazione finora
- Surgical Procedures in The Intensive Care Unit - A Critical Review. (BM Dennis, OL Gunter OA CC 2013)Documento7 pagineSurgical Procedures in The Intensive Care Unit - A Critical Review. (BM Dennis, OL Gunter OA CC 2013)PkernNessuna valutazione finora
- Spinning Dials - How To Dominate The Ventilator - Handout (EMCRIT, ARDSnet) PDFDocumento5 pagineSpinning Dials - How To Dominate The Ventilator - Handout (EMCRIT, ARDSnet) PDFPkernNessuna valutazione finora
- Rule of 4 Illustration Brainstem (LITFL)Documento1 paginaRule of 4 Illustration Brainstem (LITFL)PkernNessuna valutazione finora
- Heart Failure PDFDocumento9 pagineHeart Failure PDFPkernNessuna valutazione finora
- Renal, Fluids and Acid-Base Notes (Chris Andersen, ICUPrimaryPrep - Com)Documento8 pagineRenal, Fluids and Acid-Base Notes (Chris Andersen, ICUPrimaryPrep - Com)PkernNessuna valutazione finora
- Diuretics and Renal HormonesDocumento3 pagineDiuretics and Renal HormonesPkernNessuna valutazione finora
- Cardiac Murmurs - 1p Cheat Sheet PDFDocumento1 paginaCardiac Murmurs - 1p Cheat Sheet PDFPkernNessuna valutazione finora
- Cardiology Notes (Chris Andersen, ICUPrimaryPrep - Com) PDFDocumento16 pagineCardiology Notes (Chris Andersen, ICUPrimaryPrep - Com) PDFPkernNessuna valutazione finora
- Heart FailureDocumento9 pagineHeart FailurePkernNessuna valutazione finora
- Emergency Department Drug Card - For ALIEM - Sep 2013Documento1 paginaEmergency Department Drug Card - For ALIEM - Sep 2013PkernNessuna valutazione finora
- Neurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Documento11 pagineNeurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Pkern100% (1)
- ED Drug Treatment Cheat Sheet ANZICSDocumento1 paginaED Drug Treatment Cheat Sheet ANZICSPkernNessuna valutazione finora
- Neurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Documento11 pagineNeurology Notesc (Chris Andersen, ICUPrimaryPrep - Com)Pkern100% (1)
- 2014 CCM Review Notes Jon-Emile S. Kenny M.D, 2014Documento142 pagine2014 CCM Review Notes Jon-Emile S. Kenny M.D, 2014PkernNessuna valutazione finora
- Chapter 4 Pathophysiology of Edema FormationDocumento8 pagineChapter 4 Pathophysiology of Edema FormationPkernNessuna valutazione finora
- Anaesthetic Drugs Cheat SheetsDocumento2 pagineAnaesthetic Drugs Cheat SheetsPkern100% (3)
- Respiratory Notes (Chris Andersen, ICUPrimaryPrep - Com)Documento14 pagineRespiratory Notes (Chris Andersen, ICUPrimaryPrep - Com)PkernNessuna valutazione finora
- Chapter 3 The Lymphatic VasculatureDocumento8 pagineChapter 3 The Lymphatic VasculaturePkernNessuna valutazione finora
- ATPDDocumento11 pagineATPDsagar189Nessuna valutazione finora
- Drug Overdose: Dr. Diah Ari Safitri, SPPDDocumento9 pagineDrug Overdose: Dr. Diah Ari Safitri, SPPDmkafabillahNessuna valutazione finora
- 2016 AVA NotesDocumento7 pagine2016 AVA NotesPremNessuna valutazione finora
- Asthma Nursing Care Plans - LippincottDocumento45 pagineAsthma Nursing Care Plans - LippincottDyllanoNessuna valutazione finora
- Allergy Diagnosis Reference GuideDocumento10 pagineAllergy Diagnosis Reference GuidevanjadamjanovicNessuna valutazione finora
- Argument Essay - Kelsey OlearyDocumento8 pagineArgument Essay - Kelsey Olearyapi-280611064Nessuna valutazione finora
- Nurses' Notes: Patient Cherry Dr. MDocumento4 pagineNurses' Notes: Patient Cherry Dr. MNicxx GamingNessuna valutazione finora
- Neuroimaging Advances in Holoprosencephaly: Re Ning The Spectrum of The Midline MalformationDocumento13 pagineNeuroimaging Advances in Holoprosencephaly: Re Ning The Spectrum of The Midline Malformationfamiliesforhope100% (1)
- Stratification in The Cox Model: Patrick BrehenyDocumento20 pagineStratification in The Cox Model: Patrick BrehenyRaiJúniorNessuna valutazione finora
- The Principle of Cataract Surgery: Andari Putri WardhaniDocumento16 pagineThe Principle of Cataract Surgery: Andari Putri WardhaniAndari Putri WardhaniNessuna valutazione finora
- Lista Clsi-2014 VTKDocumento1 paginaLista Clsi-2014 VTKMessy ToapantaNessuna valutazione finora
- 11 Common Symptoms of CandidaDocumento12 pagine11 Common Symptoms of Candidasantana2013Nessuna valutazione finora
- # of The Distal RadiusDocumento98 pagine# of The Distal RadiusabhinavaiimsNessuna valutazione finora
- Mergency Medicine, Second Edition: 60 Acid-Base DisordersDocumento21 pagineMergency Medicine, Second Edition: 60 Acid-Base DisordersLukman NurfauziNessuna valutazione finora
- FMConsensusDocumentbk 1Documento105 pagineFMConsensusDocumentbk 1Cheryl BensonNessuna valutazione finora
- History of Present IllnessDocumento4 pagineHistory of Present Illnessegabe386Nessuna valutazione finora
- Horizontal Jaw RelationDocumento101 pagineHorizontal Jaw Relationruchika0% (1)
- DCFS Mandated FormDocumento1 paginaDCFS Mandated FormAshraf AhmedNessuna valutazione finora
- Phlebotomy ProblemsDocumento20 paginePhlebotomy ProblemsNatalie Enriquez100% (1)
- DMNCP - Imbalanced Nutrition Less Than Body RequirementsDocumento1 paginaDMNCP - Imbalanced Nutrition Less Than Body RequirementsMel Izhra N. MargateNessuna valutazione finora
- The Code of Ethics For NursesDocumento8 pagineThe Code of Ethics For NursesZulkifli PomalangoNessuna valutazione finora