Potrebbero piacerti anche
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5795)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Removal of Methylene Blue by Activated Carbon Prepared From Waste in A Fixed Bed ColumnDocumento9 pagineRemoval of Methylene Blue by Activated Carbon Prepared From Waste in A Fixed Bed ColumnBarryNessuna valutazione finora
- UOP GB 562 Adsorbent Data SheetDocumento2 pagineUOP GB 562 Adsorbent Data SheetrbajuadiNessuna valutazione finora
- Technip Separations PDFDocumento15 pagineTechnip Separations PDFProcess EngineerNessuna valutazione finora
- Removal of Natural Organic Matter and Arsenic From Water by Electrocoagulation/ Otation Continuous Ow ReactorDocumento8 pagineRemoval of Natural Organic Matter and Arsenic From Water by Electrocoagulation/ Otation Continuous Ow ReactorazerfazNessuna valutazione finora
- Chapter 18 Surface ChemistryDocumento53 pagineChapter 18 Surface ChemistryChicken ChickenNessuna valutazione finora
- ISO834 Fire Resistance TestsDocumento4 pagineISO834 Fire Resistance TestsVictor Hugo0% (1)
- Corrosion Inhibitors - Principles, Mechanisms and Applications PDFDocumento16 pagineCorrosion Inhibitors - Principles, Mechanisms and Applications PDFleonardoNessuna valutazione finora
- Indian Association of Chemistry Teachers: National Standard Examination in Chemistry (Nsec) 2018-19Documento27 pagineIndian Association of Chemistry Teachers: National Standard Examination in Chemistry (Nsec) 2018-19sankalp somaniNessuna valutazione finora
- SGP LLP PresentationDocumento70 pagineSGP LLP PresentationАэлитаNessuna valutazione finora
- Internal Auto Drain ValveDocumento6 pagineInternal Auto Drain ValveKisnapneumatics SMNessuna valutazione finora
- Oil Spills Cleanup Chikcken FeatherDocumento5 pagineOil Spills Cleanup Chikcken FeatherFaris MatNessuna valutazione finora
- Creep PDFDocumento236 pagineCreep PDFoshadhivNessuna valutazione finora
- FCC MANUAL 5-FCC Catalyst AnalysisDocumento11 pagineFCC MANUAL 5-FCC Catalyst AnalysisshanpyanNessuna valutazione finora
- Research Proposal: (Synopsis) 1. Area / Specialization of The Research WorkDocumento26 pagineResearch Proposal: (Synopsis) 1. Area / Specialization of The Research WorkSsahil Khan100% (3)
- Batch Adsorption Process of Metals and Anions For Remediation ofDocumento331 pagineBatch Adsorption Process of Metals and Anions For Remediation ofCesar Orlando Villalobos HipolitoNessuna valutazione finora
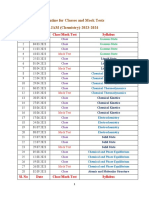
- Routine For JAM - 2023-2024Documento2 pagineRoutine For JAM - 2023-2024Rahul NathNessuna valutazione finora
- Compact Thermal Energy Storage StudyDocumento46 pagineCompact Thermal Energy Storage StudyRaif QelaNessuna valutazione finora
- ZeoliteDocumento2 pagineZeoliteNestramiNessuna valutazione finora
- Selective Hydrogen at I On of Phenol and Related DerivativesDocumento16 pagineSelective Hydrogen at I On of Phenol and Related DerivativesLuiz Rodrigo AssisNessuna valutazione finora
- Cryogenic Engineering, Production and Use of Industrial Gases, Refrigeration EngineeringDocumento6 pagineCryogenic Engineering, Production and Use of Industrial Gases, Refrigeration Engineeringdaimon_pNessuna valutazione finora
- CRE Previous Year QuestionsDocumento14 pagineCRE Previous Year QuestionsAbhishek GadhwalNessuna valutazione finora
- 2.1 Enzyme and Cell ImmobilizationDocumento40 pagine2.1 Enzyme and Cell ImmobilizationLiliana AlingodNessuna valutazione finora
- Recent Advances in Non-Precious Metal Based Electrodes For Alkaline-Water ElectrolysisDocumento61 pagineRecent Advances in Non-Precious Metal Based Electrodes For Alkaline-Water ElectrolysisShauvik BhattacharyaNessuna valutazione finora
- Topic 5 Principle of Water - Wastewater Treatment Processes-20191008090049 PDFDocumento111 pagineTopic 5 Principle of Water - Wastewater Treatment Processes-20191008090049 PDFLipQin YeoNessuna valutazione finora
- Infrared Light-Driven CO Overall Splitting at Room TemperatureDocumento14 pagineInfrared Light-Driven CO Overall Splitting at Room TemperatureRuchi MalhotraNessuna valutazione finora
- 07161Documento2 pagine07161mobilec100Nessuna valutazione finora
- Environmental Technology & Innovation: Parwathi Pillai, Swapnil Dharaskar, Sivakumar Pandian, Hitesh PanchalDocumento21 pagineEnvironmental Technology & Innovation: Parwathi Pillai, Swapnil Dharaskar, Sivakumar Pandian, Hitesh PanchalLos Parienticos Del LlanoNessuna valutazione finora
- Dew Point TurboexpanderDocumento18 pagineDew Point TurboexpanderGus Zalles ArrietaNessuna valutazione finora
- Air Pollution Control DevicesDocumento48 pagineAir Pollution Control DevicesPratyush Vaibhav75% (4)
- RHT NCCO Technology Comparison 2018Documento2 pagineRHT NCCO Technology Comparison 2018WilmanNessuna valutazione finora