Potrebbero piacerti anche
- Dispensacion de Medicamentos Farm y Productos Afines - 3ero DiaDocumento11 pagineDispensacion de Medicamentos Farm y Productos Afines - 3ero Diajose luis100% (1)
- Poes BoticaDocumento47 paginePoes BoticaManuel Castro Gonzales85% (39)
- Manual de Procedimientos Botica - PeruDocumento22 pagineManual de Procedimientos Botica - PeruVladimir Flores Cayllahua100% (1)
- BUENAS PRACTICAS DE ALMACENAMIENTO - Doc MargaritaDocumento76 pagineBUENAS PRACTICAS DE ALMACENAMIENTO - Doc MargaritaArmor24100% (1)
- Manual de Buenas Practicas de AlmacenamientoDocumento6 pagineManual de Buenas Practicas de AlmacenamientoclaudiaNessuna valutazione finora
- Qué Son Las BPM y BPADocumento6 pagineQué Son Las BPM y BPAGerson Sanchez GonzalesNessuna valutazione finora
- Buenas Prácticas de Almacenamiento de Productos Farmacéuticos y AfinesDocumento6 pagineBuenas Prácticas de Almacenamiento de Productos Farmacéuticos y AfinesMayner GaltNessuna valutazione finora
- Recepción de medicamentos e insumos médicos en farmacia comunitariaDocumento9 pagineRecepción de medicamentos e insumos médicos en farmacia comunitariaNenaEdickaNessuna valutazione finora
- EvaluacionDocumento17 pagineEvaluacionpatty tsNessuna valutazione finora
- PROC-004 PROCEDIMIENTO OPERATIVO DE RECEPCION Y EL ANALISIS ORGANOLEPTICO. Rev 0Documento4 paginePROC-004 PROCEDIMIENTO OPERATIVO DE RECEPCION Y EL ANALISIS ORGANOLEPTICO. Rev 0Lio MarinNessuna valutazione finora
- PoeDocumento8 paginePoeChuquillanquiRamosBenedictaNessuna valutazione finora
- De Los Envases, Etiquetas, RotulosDocumento24 pagineDe Los Envases, Etiquetas, RotulosErira Juan100% (3)
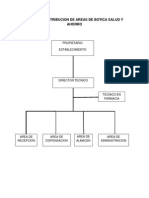
- Salud y Ahorro CORREGIRDocumento20 pagineSalud y Ahorro CORREGIRAlizon Valencia ChuraNessuna valutazione finora
- Recepcion Tecnica y AdministrativaDocumento10 pagineRecepcion Tecnica y Administrativaelvia CalampaNessuna valutazione finora
- Grupos FarmacológicosDocumento54 pagineGrupos Farmacológicoskatty211250% (2)
- BPA: Buenas prácticas de almacenamiento farmacéuticoDocumento48 pagineBPA: Buenas prácticas de almacenamiento farmacéuticoDiego MendfiNessuna valutazione finora
- PROCEDIMIENTOS PARA LA RECEPCION DE LOS PRODUCTOS FARMACÉUTICOS - SantiagoDocumento21 paginePROCEDIMIENTOS PARA LA RECEPCION DE LOS PRODUCTOS FARMACÉUTICOS - SantiagoDavid Pawqar100% (3)
- MATERIAS PRIMAS E INSUMOS UTILIZADOS EN LA PREPARACION DispositivasDocumento33 pagineMATERIAS PRIMAS E INSUMOS UTILIZADOS EN LA PREPARACION DispositivasMarìaFernanda Meza100% (3)
- Procedimientos operativos estándar botica biofarmaDocumento22 pagineProcedimientos operativos estándar botica biofarmaVane PonsNessuna valutazione finora
- Mpo Botica Maria EstherDocumento38 pagineMpo Botica Maria EstherAnonymous MunIEkQ6hNessuna valutazione finora
- AlmacenamientoFarmacéuticoDocumento3 pagineAlmacenamientoFarmacéuticoYefersonNessuna valutazione finora
- Taller Decreto 677 Etiquetas, Rótulos y Empaques de Los MedicamentosDocumento4 pagineTaller Decreto 677 Etiquetas, Rótulos y Empaques de Los MedicamentosNicoll PedrazaNessuna valutazione finora
- Pruebas a los envases farmacéuticos y alimenticiosDocumento44 paginePruebas a los envases farmacéuticos y alimenticiosMarisol MontañoNessuna valutazione finora
- 4.1 Legislacion y Normas Sobre Envase, Empaque y Embalaje.Documento11 pagine4.1 Legislacion y Normas Sobre Envase, Empaque y Embalaje.Vejero Hernández Abraham UlisesNessuna valutazione finora
- Identificación de medicamentos fraudulentosDocumento28 pagineIdentificación de medicamentos fraudulentosNataly MuñozNessuna valutazione finora
- POEs Botica 2Documento20 paginePOEs Botica 2Giancarlo AlvarezNessuna valutazione finora
- Administracion de FarmaciasDocumento38 pagineAdministracion de FarmaciasJose Felipe Perez Aviles100% (3)
- Poes 1 BoticasDocumento51 paginePoes 1 BoticasYanina Mendoza Mestanza100% (2)
- Buenas Prácticas de Almacenamiento de Productos Farmacéuticos y AfinesDocumento27 pagineBuenas Prácticas de Almacenamiento de Productos Farmacéuticos y AfinesCatalina ViaderoNessuna valutazione finora
- Clase 6 Auditoria de FarmaciaDocumento3 pagineClase 6 Auditoria de Farmaciaciber jfNessuna valutazione finora
- Recepcion Tecnica y AdministrativaDocumento11 pagineRecepcion Tecnica y AdministrativaJASMINNessuna valutazione finora
- Botica Rosa de America Rectualizado 2014Documento35 pagineBotica Rosa de America Rectualizado 2014Davids QVNessuna valutazione finora
- Poes Servicio de Farmacia HWCVDocumento28 paginePoes Servicio de Farmacia HWCVKeny Elvis0% (1)
- Instrumento de EvaluaciónDocumento24 pagineInstrumento de EvaluaciónMatias HanielNessuna valutazione finora
- Taller 11Documento4 pagineTaller 11beiby cleofe salazar caicedoNessuna valutazione finora
- Requisitos Rs SteviaDocumento9 pagineRequisitos Rs SteviaFederico TAPIA JARANessuna valutazione finora
- NMX F 433 1982 PDFDocumento5 pagineNMX F 433 1982 PDFMayra Cortés GutiérrezNessuna valutazione finora
- Dokumen - Tips Poes-BoticaDocumento47 pagineDokumen - Tips Poes-Boticajanethchaponansantisteban1Nessuna valutazione finora
- Buenas Practicas de Almacenamiento (Bpa) : Química Farmacéutica Universidad Nacional de ColombiaDocumento33 pagineBuenas Practicas de Almacenamiento (Bpa) : Química Farmacéutica Universidad Nacional de ColombiaMelvi AlarconNessuna valutazione finora
- 1 Programa de Gestion de Medicamentos, Dispositivos Médicos e Insumos Jorge MartinezDocumento7 pagine1 Programa de Gestion de Medicamentos, Dispositivos Médicos e Insumos Jorge MartinezDiana Pat HenaoNessuna valutazione finora
- Recepción de medicamentos y materiales médicos en centro navalDocumento8 pagineRecepción de medicamentos y materiales médicos en centro navalcarlos ubillusNessuna valutazione finora
- Manual de Buenas Practicas de AlmacenamientoDocumento11 pagineManual de Buenas Practicas de AlmacenamientoClaudia NolazcoNessuna valutazione finora
- Protocolo Pedido y Recepcion de MedicamentosDocumento14 pagineProtocolo Pedido y Recepcion de MedicamentosNancyNessuna valutazione finora
- FarmaciaDocumento4 pagineFarmaciaJhosias Abanto DavilaNessuna valutazione finora
- D.S. 016-2011-SA I y IIDocumento11 pagineD.S. 016-2011-SA I y IImayrin torres castanedaNessuna valutazione finora
- 1 Guía Práctica de Elaboración de Fichas Técnicas de Alimentos - FRUTAS Y VERDURASDocumento9 pagine1 Guía Práctica de Elaboración de Fichas Técnicas de Alimentos - FRUTAS Y VERDURASFiorella Anghelina DlcNessuna valutazione finora
- Karlita 1Documento29 pagineKarlita 1karla0noaNessuna valutazione finora
- Manual de Procesos y ProcedimientosDocumento43 pagineManual de Procesos y ProcedimientosMarthaNievesPeregrinoNessuna valutazione finora
- Normatividad de Productos Fitoterapéuticos en Colombia AdjDocumento17 pagineNormatividad de Productos Fitoterapéuticos en Colombia AdjZoraya MartinezNessuna valutazione finora
- Paso 1 Fundamentacion Lina TrejosDocumento4 paginePaso 1 Fundamentacion Lina TrejosCECILIA MUÑOZ100% (1)
- Solicitud de Autorizacin Sanitaria de Envases y Empaques para AlimentosDocumento2 pagineSolicitud de Autorizacin Sanitaria de Envases y Empaques para AlimentosGato de TejadoNessuna valutazione finora
- Solicitud de Autorizacin Sanitaria de Envases y Empaques para Alimentos PDFDocumento2 pagineSolicitud de Autorizacin Sanitaria de Envases y Empaques para Alimentos PDFYorvic GodoyNessuna valutazione finora
- Buenas Practicas de Almacenamiento de Medicamentos e Insumos Medicos.Documento52 pagineBuenas Practicas de Almacenamiento de Medicamentos e Insumos Medicos.Gabriel ParraNessuna valutazione finora
- Duraznos en Almíbar NMX-F-034-1982Documento5 pagineDuraznos en Almíbar NMX-F-034-1982oscarNessuna valutazione finora
- Norma Tecnica Sanitaria Aplicable A Los Azucares y Jarabes Destinados Al Consumo HumanoDocumento16 pagineNorma Tecnica Sanitaria Aplicable A Los Azucares y Jarabes Destinados Al Consumo HumanoLuiggi G Quispe RNessuna valutazione finora
- Resumen Ley CosmeticosDocumento10 pagineResumen Ley CosmeticosROCIO BETHANCOURTNessuna valutazione finora
- Taller Decreto 677 Rotulos Empaques y EtiquetasDocumento5 pagineTaller Decreto 677 Rotulos Empaques y EtiquetasCristian David50% (4)
- Adm y Leg Boticas 1Documento136 pagineAdm y Leg Boticas 1Joe GuevaraNessuna valutazione finora
- Sal Farm Requisitos060205021Documento44 pagineSal Farm Requisitos060205021JuanPabloGalvagnoAhubanNessuna valutazione finora
- Supervisión de las operaciones preliminares y técnicas de manipulación. HOTR0110Da EverandSupervisión de las operaciones preliminares y técnicas de manipulación. HOTR0110Nessuna valutazione finora
- Intoxicaciones y AntidotosDocumento1 paginaIntoxicaciones y AntidotosYuli Katherine MaldonadoNessuna valutazione finora
- Historia CriminalísticaDocumento75 pagineHistoria CriminalísticaJosafat Vermillion75% (4)
- Sesion Educativa de ParasitosisDocumento10 pagineSesion Educativa de ParasitosisYuli Katherine MaldonadoNessuna valutazione finora
- MonografiaDocumento24 pagineMonografiaYuli Katherine MaldonadoNessuna valutazione finora
- Compresibilidad en La Medición de Velocidad de Aire Con Tubos de PitotDocumento4 pagineCompresibilidad en La Medición de Velocidad de Aire Con Tubos de PitotDavid YúgarNessuna valutazione finora
- Matriz Evaluativa Tridimensional 8° BásicoDocumento4 pagineMatriz Evaluativa Tridimensional 8° BásicoFrancisco B. CatNessuna valutazione finora
- MT TuberiasDocumento112 pagineMT TuberiasJaime Alejandro HerreraNessuna valutazione finora
- Dimensionamiento de LotesDocumento23 pagineDimensionamiento de LotesJhon RamirezNessuna valutazione finora
- Caso-Palma Africana Kukra HillDocumento40 pagineCaso-Palma Africana Kukra HillGabriel RodriguezNessuna valutazione finora
- Lagunas Aireadas MecanicamenteDocumento31 pagineLagunas Aireadas Mecanicamentefredy.1rt@gmail.comNessuna valutazione finora
- Biología: André Alexander Quintal RubioDocumento4 pagineBiología: André Alexander Quintal RubioANDRE ALEXANDER QUINTAL RUBIONessuna valutazione finora
- Diseño de IncineradoresDocumento80 pagineDiseño de IncineradoresKen Rojas FuentesNessuna valutazione finora
- Ensayo de Expansion de Los SuelosDocumento24 pagineEnsayo de Expansion de Los Suelosoriana12100% (2)
- DISEÑO Y CONSTRUCCION DE EDIFICIOS CAP VII, FLEXO-COMPRESIÓN BIAXIAL S. Vinnakota PDFDocumento47 pagineDISEÑO Y CONSTRUCCION DE EDIFICIOS CAP VII, FLEXO-COMPRESIÓN BIAXIAL S. Vinnakota PDFVictor CarrionNessuna valutazione finora
- Sistemas Estructurales Integrados PDFDocumento6 pagineSistemas Estructurales Integrados PDFINGENIERIA COLOMBIANANessuna valutazione finora
- Presentacion Lodos ActivadosDocumento14 paginePresentacion Lodos ActivadosElkin Andres Guzman0% (1)
- Resistencia de MaterialesDocumento9 pagineResistencia de MaterialesWill FebresNessuna valutazione finora
- Sesión 4 y 5 - Estructura Atómica 1Documento13 pagineSesión 4 y 5 - Estructura Atómica 1Maria Alejandra Ortiz ZapataNessuna valutazione finora
- Calidad IntralaboratorioDocumento6 pagineCalidad IntralaboratorioeduardoNessuna valutazione finora
- Ejercicios Equilibrio QuímicoDocumento5 pagineEjercicios Equilibrio QuímicofaltriqueraNessuna valutazione finora
- Informe Laboratorio 2 Quimica UniDocumento11 pagineInforme Laboratorio 2 Quimica UniLeiver Raul RCNessuna valutazione finora
- Concentrados Herbicidas Acuosos 2859612T3Documento19 pagineConcentrados Herbicidas Acuosos 2859612T3Alfredo MéndezNessuna valutazione finora
- Trac HeliumDocumento1 paginaTrac HeliumCarlos gongoraNessuna valutazione finora
- Tratamiento Termico LaboratorioDocumento18 pagineTratamiento Termico LaboratoriojhoanNessuna valutazione finora
- Biofisica Medica Unt ExamenDocumento1 paginaBiofisica Medica Unt ExamenZavala H IrvingNessuna valutazione finora
- Quim T3 2022Documento3 pagineQuim T3 2022Ainara Caro RaceroNessuna valutazione finora
- Practica 9 AdsorcionDocumento9 paginePractica 9 AdsorcionAna WongNessuna valutazione finora
- Tarea # 1Documento12 pagineTarea # 1Vivian VásquezNessuna valutazione finora
- Calculos de Sistemas VaporDocumento11 pagineCalculos de Sistemas VaporJulio Mamani100% (1)
- Recursos Naturales Renovables y No RenovablesDocumento11 pagineRecursos Naturales Renovables y No RenovablesDeygui Jiset MuñozNessuna valutazione finora
- Historia y evolución de los biomaterialesDocumento13 pagineHistoria y evolución de los biomaterialesAnnelle PalafoxNessuna valutazione finora
- M-259 VE MS5 S5000 Con H5 (Rev.1) PDFDocumento18 pagineM-259 VE MS5 S5000 Con H5 (Rev.1) PDFAnonymous YmXY1bcNessuna valutazione finora
- Informe Final Grupo 4Documento17 pagineInforme Final Grupo 4KEVIN ISRAEL GUILLEN MOLLINEDONessuna valutazione finora
- INFORMEDocumento18 pagineINFORMEFranz Gustavo Vargas MamaniNessuna valutazione finora