Potrebbero piacerti anche
- Syncope E:Manag Afp (Dragged) 2Documento1 paginaSyncope E:Manag Afp (Dragged) 2M.DalaniNessuna valutazione finora
- CEP BPSD Discussion Guide ENG RFCG Updated2019 PDFDocumento8 pagineCEP BPSD Discussion Guide ENG RFCG Updated2019 PDFM.DalaniNessuna valutazione finora
- CMPA MR - Diagnostic - Tips-E (Dragged)Documento1 paginaCMPA MR - Diagnostic - Tips-E (Dragged)M.DalaniNessuna valutazione finora
- CMPA MR - Diagnostic - Tips-E (Dragged) 2Documento1 paginaCMPA MR - Diagnostic - Tips-E (Dragged) 2M.DalaniNessuna valutazione finora
- Syncope E:Manag Afp (Dragged) 3Documento1 paginaSyncope E:Manag Afp (Dragged) 3M.DalaniNessuna valutazione finora
- Baseline Functional Assessment Tool - Draft - 6Documento2 pagineBaseline Functional Assessment Tool - Draft - 6M.DalaniNessuna valutazione finora
- Bulimia Nervosa:: Purging Patients' and Families' MisconceptionsDocumento17 pagineBulimia Nervosa:: Purging Patients' and Families' MisconceptionsM.DalaniNessuna valutazione finora
- Canadian Approach Assisted Dying eDocumento19 pagineCanadian Approach Assisted Dying eM.DalaniNessuna valutazione finora
- Bowel Care: Cancer Care Ontario's Symptom Management Guide-to-PracticeDocumento27 pagineBowel Care: Cancer Care Ontario's Symptom Management Guide-to-PracticeM.DalaniNessuna valutazione finora
- Assessment Using Acronym O, P, Q, R, S, T, U and V: Algorithm Dysgeusia in Adults With Cancer: Screening and AssessmentDocumento2 pagineAssessment Using Acronym O, P, Q, R, S, T, U and V: Algorithm Dysgeusia in Adults With Cancer: Screening and AssessmentM.DalaniNessuna valutazione finora
- Making It Happen: TogetherDocumento12 pagineMaking It Happen: TogetherM.DalaniNessuna valutazione finora
- Mccqe Objective ListDocumento4 pagineMccqe Objective ListM.DalaniNessuna valutazione finora
- 10 Steps To Better Prognostication Table CopyrightDocumento1 pagina10 Steps To Better Prognostication Table CopyrightM.DalaniNessuna valutazione finora
- Neuro Notes UWDocumento91 pagineNeuro Notes UWM.DalaniNessuna valutazione finora
- Cancer Care Ontario's Symptom Management Guide-to-Practice: Loss of AppetiteDocumento31 pagineCancer Care Ontario's Symptom Management Guide-to-Practice: Loss of AppetiteM.DalaniNessuna valutazione finora
- 99 Topics StudynotesDocumento138 pagine99 Topics StudynotesM.Dalani100% (1)
- Guideline: Preoperative Medication ManagementDocumento18 pagineGuideline: Preoperative Medication ManagementM.DalaniNessuna valutazione finora
- Bowel Loops and Eyelid Droops CaseDocumento3 pagineBowel Loops and Eyelid Droops CaseM.DalaniNessuna valutazione finora
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Endodontic: EmergenciesDocumento53 pagineEndodontic: EmergenciesAhmed S. Abu-Haimed100% (1)
- Biology - Unit 4 Kingdom ProtistaDocumento0 pagineBiology - Unit 4 Kingdom Protistawww.bhawesh.com.npNessuna valutazione finora
- Vaccines For COVID-19 - The Current State of Play PDFDocumento8 pagineVaccines For COVID-19 - The Current State of Play PDFdiana.alyNessuna valutazione finora
- Vaccine Safety E Course ManualDocumento207 pagineVaccine Safety E Course ManualDita TrastitaNessuna valutazione finora
- Non Sirosis PHDocumento11 pagineNon Sirosis PHHIstoryNessuna valutazione finora
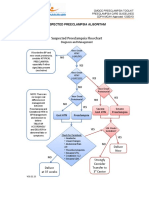
- Suspected Preeclampsia AlgorithmDocumento1 paginaSuspected Preeclampsia Algorithmmisstina.19876007100% (1)
- Nursing Care of A Child With Respiratory DisorderDocumento69 pagineNursing Care of A Child With Respiratory DisorderTamil Villardo100% (1)
- Essential, Trace, and Nonessential Ions-1Documento3 pagineEssential, Trace, and Nonessential Ions-1John AndanNessuna valutazione finora
- Case StudyDocumento12 pagineCase Studyapi-291857811Nessuna valutazione finora
- Sexually Transmitted Diseases OverviewDocumento45 pagineSexually Transmitted Diseases OverviewJean De Vera MelendezNessuna valutazione finora
- 15 - Chapter 5 PDFDocumento140 pagine15 - Chapter 5 PDFHimanshu YadavNessuna valutazione finora
- Respiratory Distress of The New BornDocumento15 pagineRespiratory Distress of The New BornJicko Street HooligansNessuna valutazione finora
- Parts and LotsDocumento5 pagineParts and LotsAnahiti Atena100% (1)
- 15 - Minute Test (1) : Exercise 1. Find The Word Which Has A Different Sound in The Part UnderlinedDocumento2 pagine15 - Minute Test (1) : Exercise 1. Find The Word Which Has A Different Sound in The Part UnderlinedPhương Anh NguyễnNessuna valutazione finora
- Method SujokDocumento4 pagineMethod Sujokbhupatin100% (6)
- Neonatal SepsisDocumento6 pagineNeonatal SepsisAisyah ShawtyNessuna valutazione finora
- Diseases of The Esophagus DR Lapuz MCDDocumento6 pagineDiseases of The Esophagus DR Lapuz MCDMiguel Cuevas Dolot100% (1)
- OBGYN Shelf NotesDocumento7 pagineOBGYN Shelf NotesaelteeNessuna valutazione finora
- Allergic Rhinitis: Practical Management StrategiesDocumento6 pagineAllergic Rhinitis: Practical Management StrategiesSravan KrishnaNessuna valutazione finora
- Diet Survey (SUKRITI)Documento37 pagineDiet Survey (SUKRITI)Juhi Neogi100% (1)
- Medical Surgical Nursing Guillain Barre SyndromeDocumento6 pagineMedical Surgical Nursing Guillain Barre SyndromeNoelyn BaluyanNessuna valutazione finora
- MS Sas 5Documento5 pagineMS Sas 5rereNessuna valutazione finora
- CROUPDocumento26 pagineCROUPRahul MehtaNessuna valutazione finora
- ME/CFS Breathing TechniquesDocumento1 paginaME/CFS Breathing TechniquesCheryl BensonNessuna valutazione finora
- Respiratory System-Review PathoDocumento100 pagineRespiratory System-Review PathoSadiePartington-RiopelleNessuna valutazione finora
- Annals of Medicine and Surgery: Muhammad Anis Baskara, Firdian Makrufardi, Ardiana DinisariDocumento4 pagineAnnals of Medicine and Surgery: Muhammad Anis Baskara, Firdian Makrufardi, Ardiana Dinisarialfan MaulanaNessuna valutazione finora
- EB-160 Cupping Therapy Encyclopedia - Tamer Shaban-1 PDFDocumento225 pagineEB-160 Cupping Therapy Encyclopedia - Tamer Shaban-1 PDFTanuku Net60% (5)
- PhysicalEducation SQP PDFDocumento7 paginePhysicalEducation SQP PDFAman JaiswalNessuna valutazione finora
- Yoga For Better HealthDocumento39 pagineYoga For Better HealthCservenák GáborNessuna valutazione finora
- DynaMed Plus - Pulmonary Embolism (PE)Documento85 pagineDynaMed Plus - Pulmonary Embolism (PE)Gamer MadaNessuna valutazione finora