Potrebbero piacerti anche
- Portaria #121Documento6 paginePortaria #121Giovanni SilvaNessuna valutazione finora
- 1 - Antibioticos - MD2Documento13 pagine1 - Antibioticos - MD2Filipe SehnNessuna valutazione finora
- PT - GM - 1559 RegulaçãoDocumento5 paginePT - GM - 1559 Regulaçãole_marcela8275Nessuna valutazione finora
- U - PT MS Sas 650 - 051011Documento11 pagineU - PT MS Sas 650 - 051011Regileudo Gama da SilvaNessuna valutazione finora
- Apostila RMNDocumento20 pagineApostila RMNAnna Paula Castro TeixeiraNessuna valutazione finora
- Virologia básica e clínicaDocumento45 pagineVirologia básica e clínicaRegileudo Gama da SilvaNessuna valutazione finora
- As Diferenças Entre Os Serviços de Saúde Da Alemanha e Do CanadáDocumento57 pagineAs Diferenças Entre Os Serviços de Saúde Da Alemanha e Do CanadáRegileudo Gama da SilvaNessuna valutazione finora
- CIT Resolucao 4Documento30 pagineCIT Resolucao 4Regileudo Gama da SilvaNessuna valutazione finora
- Percival Pugina - A Tomada Do Brasil Pelos Maus Brasileiros PDFDocumento296 paginePercival Pugina - A Tomada Do Brasil Pelos Maus Brasileiros PDFRamonNessuna valutazione finora
- Apostila Prática - Tecnologia Farmacêutica 2012-02Documento10 pagineApostila Prática - Tecnologia Farmacêutica 2012-02Mariana Garvil100% (1)
- Monografia FATCH 2007Documento107 pagineMonografia FATCH 2007Osvaldo Olimpio de MacedoNessuna valutazione finora
- Tipos de Evisao de Literatura PDFDocumento9 pagineTipos de Evisao de Literatura PDFClaudiaCacauNessuna valutazione finora
- (Grego) Gramática de Grego KoinêDocumento138 pagine(Grego) Gramática de Grego Koinêrmdamasio100% (39)
- A Química Quântica Na Compreensão de Teorias de Química OrgânicaDocumento5 pagineA Química Quântica Na Compreensão de Teorias de Química OrgânicaFernando Silva BetimNessuna valutazione finora
- Desafios Da Indústria Farmacêutica BrasileiraDocumento4 pagineDesafios Da Indústria Farmacêutica BrasileiraRegileudo Gama da SilvaNessuna valutazione finora
- RMN 1H e 13C da Química OrgânicaDocumento17 pagineRMN 1H e 13C da Química OrgânicaAgnes Mih RibeiroNessuna valutazione finora
- V 84 N 5 A 03Documento9 pagineV 84 N 5 A 03Regileudo Gama da SilvaNessuna valutazione finora
- Banner APS Talassemias PDFDocumento1 paginaBanner APS Talassemias PDFRegileudo Gama da SilvaNessuna valutazione finora
- Art 07 Dieta ProteínasDocumento8 pagineArt 07 Dieta ProteínasRegileudo Gama da SilvaNessuna valutazione finora
- Conselho Nacional de Saúde Próximas ReuniõesDocumento1 paginaConselho Nacional de Saúde Próximas ReuniõesRegileudo Gama da SilvaNessuna valutazione finora
- Farmacopéia Brasileira 5 Ed. Volume 1 - AnvisaDocumento545 pagineFarmacopéia Brasileira 5 Ed. Volume 1 - AnvisaLuizSantos100% (3)
- Apostila 7 (Pic-2008) - CriptografiaDocumento225 pagineApostila 7 (Pic-2008) - Criptografiadgoiana732Nessuna valutazione finora
- Formulario Hospitalar Nacional de MedicamentosDocumento255 pagineFormulario Hospitalar Nacional de MedicamentosTelmo AlexandreNessuna valutazione finora
- Química Farmacêutica - Capa MódulosDocumento1 paginaQuímica Farmacêutica - Capa MódulosLeticia BossoloNessuna valutazione finora
- Resolução Da Diretoria Colegiada - RDC Nº 214, de 12 de Dezembro de 2006. ( )Documento1 paginaResolução Da Diretoria Colegiada - RDC Nº 214, de 12 de Dezembro de 2006. ( )Regileudo Gama da SilvaNessuna valutazione finora
- A Resposta Reformada Aos Apelos Bíblicos Do PentecostalismoDocumento12 pagineA Resposta Reformada Aos Apelos Bíblicos Do PentecostalismoRegileudo Gama da SilvaNessuna valutazione finora
- Geometria analítica: retas e ângulosDocumento3 pagineGeometria analítica: retas e ângulosJulio CarraroNessuna valutazione finora
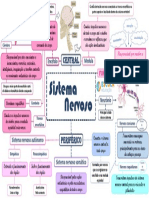
- Sistema nervoso: estrutura e funções principaisDocumento1 paginaSistema nervoso: estrutura e funções principaisGabriela PiresNessuna valutazione finora
- 2o Encontro Regional História MídiaDocumento981 pagine2o Encontro Regional História MídiaRodrigoNessuna valutazione finora
- Nova espécie de Jupoata da Costa RicaDocumento3 pagineNova espécie de Jupoata da Costa RicaSimone BM MinduimNessuna valutazione finora
- Redação Suspensão A ArDocumento1 paginaRedação Suspensão A ArMateus FerreiraNessuna valutazione finora
- Como entender a BíbliaDocumento30 pagineComo entender a BíbliaRicardo NojosaNessuna valutazione finora
- Partilha de Imovel em IRC e No Socio PT18751 1 de Março2017Documento14 paginePartilha de Imovel em IRC e No Socio PT18751 1 de Março2017Americo AraujoNessuna valutazione finora
- Relatório 2 - Movimento Sobre Um Trilho de Ar HorizontalDocumento11 pagineRelatório 2 - Movimento Sobre Um Trilho de Ar HorizontalAna CostaNessuna valutazione finora
- Direito a 1/3 hora-atividade para todos os profissionais da educaçãoDocumento7 pagineDireito a 1/3 hora-atividade para todos os profissionais da educaçãoTiago TondinelliNessuna valutazione finora
- Análise de riscos de escavaçãoDocumento5 pagineAnálise de riscos de escavaçãoAna Carolina NascimentoNessuna valutazione finora
- Rolando Nassau - Ambientes Do Culto IIDocumento3 pagineRolando Nassau - Ambientes Do Culto IIIVANTEOLOGIANessuna valutazione finora
- Cartilha Informativa - Síndrome de BurnoutDocumento10 pagineCartilha Informativa - Síndrome de BurnoutIsabela SouzaNessuna valutazione finora
- Livro SenhoraDocumento12 pagineLivro SenhoraSamuel FiaisNessuna valutazione finora
- Nema11 Manual U1 Res PDFDocumento59 pagineNema11 Manual U1 Res PDFJaime FonsecaNessuna valutazione finora
- Manual de Plantas ComestiveisDocumento15 pagineManual de Plantas ComestiveisElieser SantosNessuna valutazione finora
- As quatro sugestões para mudançaDocumento3 pagineAs quatro sugestões para mudançaRegina CoeliNessuna valutazione finora
- (Da Dor Ao Alívio) Ebook 3Documento16 pagine(Da Dor Ao Alívio) Ebook 3kaoliveNessuna valutazione finora
- Risoto de Frango - Cozinha TécnicaDocumento3 pagineRisoto de Frango - Cozinha TécnicaCasa CasaNessuna valutazione finora
- A importância da psicomotricidade no desenvolvimento infantilDocumento52 pagineA importância da psicomotricidade no desenvolvimento infantilSandro Farias100% (4)
- Eixo Tematico PreconceitoDocumento2 pagineEixo Tematico PreconceitoFelipe CavalcanteNessuna valutazione finora
- Macrodantina PDFDocumento3 pagineMacrodantina PDFDiego FelixNessuna valutazione finora
- Tratamentos térmicos em aço médio carbonoDocumento26 pagineTratamentos térmicos em aço médio carbonoLorena MeloNessuna valutazione finora
- Prova de HistoriaDocumento2 pagineProva de HistoriaBruna SantanaNessuna valutazione finora
- Lista de Geometria Circunferencias e Quadrilc3a1teros1Documento6 pagineLista de Geometria Circunferencias e Quadrilc3a1teros1zilmarsoares5618Nessuna valutazione finora
- Ficha de atendimento previdenciárioDocumento8 pagineFicha de atendimento previdenciárioEmanuely Lima100% (1)
- Evolução morfológica em áreas de deformação tectônicaDocumento14 pagineEvolução morfológica em áreas de deformação tectônicaGeoman BahiaNessuna valutazione finora
- Franquia Barbearia Seu Elias - Menos deDocumento21 pagineFranquia Barbearia Seu Elias - Menos deDaniel RodriguesNessuna valutazione finora
- Doenças Crônicas Que Determinam A Realização de Atividades Home OfficeDocumento6 pagineDoenças Crônicas Que Determinam A Realização de Atividades Home OfficeAline Sousa JorgeNessuna valutazione finora
- 38fatorial, Permutações E Arranjos SimplesDocumento40 pagine38fatorial, Permutações E Arranjos SimplesZé OtávioNessuna valutazione finora
- Crimes em Especie Unidade 3Documento16 pagineCrimes em Especie Unidade 3clichardson hipolitoNessuna valutazione finora
- Mente calma: Técnicas para controlar pensamentos intrusivosDa EverandMente calma: Técnicas para controlar pensamentos intrusivosValutazione: 4 su 5 stelle4/5 (6)
- Manual das Microexpressões: Há informações que o rosto não escondeDa EverandManual das Microexpressões: Há informações que o rosto não escondeValutazione: 4.5 su 5 stelle4.5/5 (4)
- Minuto da gratidão: O desafio dos 90 dias que mudará a sua vidaDa EverandMinuto da gratidão: O desafio dos 90 dias que mudará a sua vidaValutazione: 5 su 5 stelle5/5 (8)
- 35 Técnicas e Curiosidades Mentais: Porque a mente também deve evoluirDa Everand35 Técnicas e Curiosidades Mentais: Porque a mente também deve evoluirValutazione: 5 su 5 stelle5/5 (3)
- Freud no século XXI: Volume 1: O que é psicanálise?Da EverandFreud no século XXI: Volume 1: O que é psicanálise?Nessuna valutazione finora
- Como Analisar as Pessoas. Linguagem CorporalDa EverandComo Analisar as Pessoas. Linguagem CorporalValutazione: 5 su 5 stelle5/5 (4)
- Focar: Supere a procrastinação e aumente a força de vontade e a atençãoDa EverandFocar: Supere a procrastinação e aumente a força de vontade e a atençãoValutazione: 4.5 su 5 stelle4.5/5 (53)
- Os Códigos do Mindset da Prosperidade: destrave os bloqueios em sua mente e cresça em todos os aspectos de sua vidaDa EverandOs Códigos do Mindset da Prosperidade: destrave os bloqueios em sua mente e cresça em todos os aspectos de sua vidaNessuna valutazione finora
- Encontre seu propósito: Como traçar um caminho em direção às suas paixões, fortalezas e autodescobertaDa EverandEncontre seu propósito: Como traçar um caminho em direção às suas paixões, fortalezas e autodescobertaValutazione: 5 su 5 stelle5/5 (7)
- Avaliação psicológica e desenvolvimento humano: Casos clínicosDa EverandAvaliação psicológica e desenvolvimento humano: Casos clínicosNessuna valutazione finora
- Treinamento cerebral: Como funcionam a inteligência e o pensamento cognitivo (2 em 1)Da EverandTreinamento cerebral: Como funcionam a inteligência e o pensamento cognitivo (2 em 1)Valutazione: 4.5 su 5 stelle4.5/5 (29)
- Psiquiatria e Jesus: transforme suas emoções em 30 diasDa EverandPsiquiatria e Jesus: transforme suas emoções em 30 diasValutazione: 5 su 5 stelle5/5 (1)
- Lei da atração: O significado da vida e atrair o que você desejaDa EverandLei da atração: O significado da vida e atrair o que você desejaValutazione: 4.5 su 5 stelle4.5/5 (22)
- Cérebro Singular: Como estimular crianças no espectro autista ou com atrasos no desenvolvimentoDa EverandCérebro Singular: Como estimular crianças no espectro autista ou com atrasos no desenvolvimentoValutazione: 5 su 5 stelle5/5 (1)