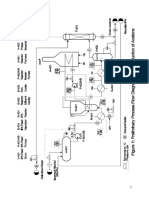
Potrebbero piacerti anche
- TOPIC: Acetic Acid Production Through Methanol Carbonylation Route Group MembersDocumento3 pagineTOPIC: Acetic Acid Production Through Methanol Carbonylation Route Group MembersThrese AreolaNessuna valutazione finora
- Acido AceticoDocumento13 pagineAcido Aceticoting_tatNessuna valutazione finora
- Acetic Acid PDFDocumento12 pagineAcetic Acid PDFhazimraadNessuna valutazione finora
- Methane Oxidation To Acetic AcidDocumento31 pagineMethane Oxidation To Acetic AcidАндрей КолесниковNessuna valutazione finora
- Chapter VIII Process and Economic OverviewDocumento38 pagineChapter VIII Process and Economic OverviewWisnu Rochman HidayatullahNessuna valutazione finora
- Cativa Process PDFDocumento12 pagineCativa Process PDFMonimNessuna valutazione finora
- Group Acetic Acid PresentationDocumento24 pagineGroup Acetic Acid PresentationNatko47Nessuna valutazione finora
- Process Evaluation Research Planning Program (Acetic Acid)Documento10 pagineProcess Evaluation Research Planning Program (Acetic Acid)Wisnu Rochman Hidayatullah0% (1)
- Cativa BPDocumento12 pagineCativa BPGhaya Bani Rushaid100% (1)
- Ethyl Benzene Plant DesignDocumento45 pagineEthyl Benzene Plant DesignfaridzawiNessuna valutazione finora
- Acetic AcidDocumento11 pagineAcetic Acidrubesh_raja0% (2)
- Star Control - Acetic Acid ProductionDocumento3 pagineStar Control - Acetic Acid ProductionHisyamAl-MuhammadiNessuna valutazione finora
- Acetic Acid Production ProcessDocumento2 pagineAcetic Acid Production ProcessAbdullaNessuna valutazione finora
- Acetic Acid ProductionDocumento4 pagineAcetic Acid Productionfatin_mujahidahNessuna valutazione finora
- Acetic Acid ProductionDocumento6 pagineAcetic Acid Productionmmoradi55Nessuna valutazione finora
- Production of Acetic Acid by Methanol CarbonylationDocumento68 pagineProduction of Acetic Acid by Methanol CarbonylationNoman Aslam100% (5)
- Vinyl AcetateDocumento5 pagineVinyl AcetateroxetteNessuna valutazione finora
- Us 20120035390Documento18 pagineUs 20120035390sariNessuna valutazione finora
- Project ReportDocumento12 pagineProject ReportRabia SabirNessuna valutazione finora
- 64788Documento35 pagine64788ghatak2100% (1)
- Acetic Acid MainDocumento58 pagineAcetic Acid MainGopal Agarwal50% (2)
- ButadineDocumento68 pagineButadineraihonaNessuna valutazione finora
- Manufacturing Methods: 1. Methanol Carbonylation 2. Acetaldehyde Oxidation 3. Ethylene Oxidation 4. Anerobic FermentationDocumento39 pagineManufacturing Methods: 1. Methanol Carbonylation 2. Acetaldehyde Oxidation 3. Ethylene Oxidation 4. Anerobic Fermentationkumari svgNessuna valutazione finora
- Production of Acetic Acid From Methanol: Petrovietnam UniversityDocumento27 pagineProduction of Acetic Acid From Methanol: Petrovietnam UniversityVăn Bão TôNessuna valutazione finora
- Official Acrylic Acid 0712docxDocumento42 pagineOfficial Acrylic Acid 0712docxTÚ Cao Ngọc ThiệnNessuna valutazione finora
- Acetic AcidDocumento8 pagineAcetic AcidMohammedRahimNessuna valutazione finora
- Acetic AcidDocumento4 pagineAcetic AcidMohit YaduwanshiNessuna valutazione finora
- Viewcontent11 PDFDocumento54 pagineViewcontent11 PDFEr Mayur PatilNessuna valutazione finora
- Produxction of Vinyl Acetate From EthyleneDocumento9 pagineProduxction of Vinyl Acetate From EthyleneSajid AliNessuna valutazione finora
- FILE 20220921 173401 Homogeneously Catalyzed Industrial ProcessesDocumento31 pagineFILE 20220921 173401 Homogeneously Catalyzed Industrial ProcessesPham ThaoNessuna valutazione finora
- التعديل النهائي محمد احمد علي عبدالله 22130011 PDFDocumento115 pagineالتعديل النهائي محمد احمد علي عبدالله 22130011 PDFRojan Pradhan0% (1)
- AcetoneDocumento7 pagineAcetoneGeorgiana AndreeaNessuna valutazione finora
- Acetaldehyde EconomicsDocumento26 pagineAcetaldehyde EconomicsKudouNessuna valutazione finora
- Styrene Production From EthylbenzeneDocumento10 pagineStyrene Production From EthylbenzeneChegg ChemNessuna valutazione finora
- Ethylene DichlorideDocumento18 pagineEthylene DichlorideAnshumanSrivastavaNessuna valutazione finora
- Lecture 5 Acetic AcidDocumento6 pagineLecture 5 Acetic AcidYan LaksanaNessuna valutazione finora
- StyreneDocumento22 pagineStyreneMohd Masri A. Razak100% (1)
- Acetic Acid Production ReportDocumento15 pagineAcetic Acid Production ReportArya Lodha100% (1)
- Liquidphasealkylationofbenzene With Ethylene 160713071057 PDFDocumento111 pagineLiquidphasealkylationofbenzene With Ethylene 160713071057 PDFFrancesca GarciaNessuna valutazione finora
- Project Ethyl Benzene .. 2019-20 .. Jay RSDocumento100 pagineProject Ethyl Benzene .. 2019-20 .. Jay RSBhatu DevareNessuna valutazione finora
- Acrolein Project Final PDFDocumento104 pagineAcrolein Project Final PDFPankaj RanaNessuna valutazione finora
- Alfonsina DME Plant DesignDocumento12 pagineAlfonsina DME Plant Designelend1993Nessuna valutazione finora
- Butanediol and Derivatives PDFDocumento4 pagineButanediol and Derivatives PDFJaamac DhiilNessuna valutazione finora
- Acetic Acid Production Process (Ct-Aceticatm) - Technology - Chiyoda CorporationDocumento2 pagineAcetic Acid Production Process (Ct-Aceticatm) - Technology - Chiyoda CorporationFauzi Abdilah100% (1)
- Formic Acid Plant: A Brief OverviewDocumento17 pagineFormic Acid Plant: A Brief OverviewMuzzamilNessuna valutazione finora
- Simulation of Different Types of Distillation Columns Usig Aspen Plus SoftwareDocumento61 pagineSimulation of Different Types of Distillation Columns Usig Aspen Plus SoftwareShashank TiwariNessuna valutazione finora
- Production of MTBE (Methyl Tert-Butyl Ether)Documento2 pagineProduction of MTBE (Methyl Tert-Butyl Ether)Aaron SinghNessuna valutazione finora
- Mthanol ProductionDocumento61 pagineMthanol Productionvv vvNessuna valutazione finora
- A 350 Tonne Per Day Phthalic Anhydride Plant: Presentation On Plant Design ForDocumento29 pagineA 350 Tonne Per Day Phthalic Anhydride Plant: Presentation On Plant Design Forbaniya is hereNessuna valutazione finora
- Benzoic Acid Manufacturing ProcessDocumento2 pagineBenzoic Acid Manufacturing ProcessSälàám Shãnü Bhåï100% (1)
- G 1 PDFDocumento199 pagineG 1 PDFKing HenryNessuna valutazione finora
- Energy Saving of A Methyl Methacrylate Separation Process PDFDocumento11 pagineEnergy Saving of A Methyl Methacrylate Separation Process PDFClaudia CelestinoNessuna valutazione finora
- Acetaldehyde Production by Ethanol DehydrogenationDocumento9 pagineAcetaldehyde Production by Ethanol DehydrogenationHugo Gerdulli AlbertinNessuna valutazione finora
- Acetylene, the Principles of Its Generation and Use A Practical Handbook on the Production, Purification, and Subsequent Treatment of Acetylene for the Development of Light, Heat, and PowerDa EverandAcetylene, the Principles of Its Generation and Use A Practical Handbook on the Production, Purification, and Subsequent Treatment of Acetylene for the Development of Light, Heat, and PowerNessuna valutazione finora
- Adiabatic Fixed-Bed Reactors: Practical Guides in Chemical EngineeringDa EverandAdiabatic Fixed-Bed Reactors: Practical Guides in Chemical EngineeringNessuna valutazione finora
- Ion Exchange TechnologyDa EverandIon Exchange TechnologyF.C. NachodNessuna valutazione finora
- Carboxylic Ortho Acid Derivatives: Preparation and Synthetic Applications: Preparation and Synthetic ApplicationsDa EverandCarboxylic Ortho Acid Derivatives: Preparation and Synthetic Applications: Preparation and Synthetic ApplicationsNessuna valutazione finora
- Markets and Commodity Figures: Total Market Turnover StatisticsDocumento6 pagineMarkets and Commodity Figures: Total Market Turnover StatisticsTiso Blackstar GroupNessuna valutazione finora
- New Microsoft Office Word DocumentDocumento10 pagineNew Microsoft Office Word DocumentcitunairNessuna valutazione finora
- (Project Eco) Latest Semifull Half Without Part RogerDocumento35 pagine(Project Eco) Latest Semifull Half Without Part RogerRoger FernandezNessuna valutazione finora
- Handout2 Fischer CarbeneDocumento5 pagineHandout2 Fischer CarbeneMuhammad ShimaNessuna valutazione finora
- ch12 - Debt FinancingDocumento60 paginech12 - Debt Financingsongvuthy100% (1)
- EXP 2 Advance Organic ChemistryDocumento7 pagineEXP 2 Advance Organic ChemistryMay Lee100% (1)
- Central Surety vs. C.N HodgesDocumento11 pagineCentral Surety vs. C.N HodgesMirzi Olga Breech SilangNessuna valutazione finora
- Neely, Rapach, Tu and Zhou (2014)Documento48 pagineNeely, Rapach, Tu and Zhou (2014)Anonymous o4X0WqcpSNessuna valutazione finora
- Phil-Am Gen Insurance Co. Vs Ramos GR No. L-20978Documento2 paginePhil-Am Gen Insurance Co. Vs Ramos GR No. L-20978Celver Joy Gaviola TampariaNessuna valutazione finora
- TD Ameritrade Free Etf List PDFDocumento6 pagineTD Ameritrade Free Etf List PDFjjy1234Nessuna valutazione finora
- CL's Handy Formula Sheet: (Useful Formulas From Marcel Finan's FM/2 Book) Compiled by Charles Lee 8/19/2010Documento15 pagineCL's Handy Formula Sheet: (Useful Formulas From Marcel Finan's FM/2 Book) Compiled by Charles Lee 8/19/2010Dũng Hữu NguyễnNessuna valutazione finora
- Electrophilic Aromatic Substitution Rxns Practice ExamDocumento25 pagineElectrophilic Aromatic Substitution Rxns Practice ExamgizatowerNessuna valutazione finora
- Bao3403 - Tutorial Questions Session 1 - Chapter 1: A. ZeroDocumento1 paginaBao3403 - Tutorial Questions Session 1 - Chapter 1: A. Zero李佳南Nessuna valutazione finora
- R07 Statistical Concepts and Market ReturnsDocumento65 pagineR07 Statistical Concepts and Market ReturnsRaviTejaNessuna valutazione finora
- Chapter 1 - Reaction Kinetics of HeterogenousDocumento68 pagineChapter 1 - Reaction Kinetics of HeterogenousFhan Sani SeowNessuna valutazione finora
- Lecture 31Documento8 pagineLecture 31Miguel RochaNessuna valutazione finora
- Makati Development Corporation - Quantity Surveyor FunctionDocumento1 paginaMakati Development Corporation - Quantity Surveyor FunctiongeassuserscribdNessuna valutazione finora
- Midterm Exam Reviewer (Mas Malala Talaga Ang Real Exam)Documento3 pagineMidterm Exam Reviewer (Mas Malala Talaga Ang Real Exam)Ying YangNessuna valutazione finora
- Payment and Milestone SchedulesDocumento71 paginePayment and Milestone SchedulesCu Ti100% (1)
- Fabozzi Bmas8 Ch03 ImDocumento26 pagineFabozzi Bmas8 Ch03 ImDavid RouleauNessuna valutazione finora
- Lipids - FatsDocumento13 pagineLipids - FatsJohn Jill T. VillamorNessuna valutazione finora
- Relative Value Models (Feb04)Documento18 pagineRelative Value Models (Feb04)api-3763138Nessuna valutazione finora
- Chapter 03 Valuing BondsDocumento37 pagineChapter 03 Valuing BondsUmme SumaiyaNessuna valutazione finora
- 12 Chemistry Impq CH10 Haloalkanes and Haloarenes 02Documento47 pagine12 Chemistry Impq CH10 Haloalkanes and Haloarenes 02Swaroop SurendraNessuna valutazione finora
- Geotechnical Engineering LECTURE NOTESDocumento65 pagineGeotechnical Engineering LECTURE NOTESmoizm5350% (2)
- DR No WorksheetDocumento4 pagineDR No Worksheetflor-calderaro-6219Nessuna valutazione finora
- Verizon UnderwritingDocumento2 pagineVerizon UnderwritingUsama FarooqNessuna valutazione finora
- Business Finance Project - DebenturesDocumento16 pagineBusiness Finance Project - DebenturesMuhammad TalhaNessuna valutazione finora
- 2 5 Marking ScheduleDocumento6 pagine2 5 Marking Scheduleapi-218511741Nessuna valutazione finora