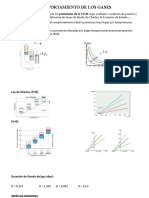
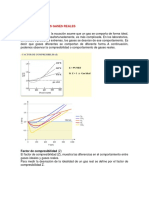
Potrebbero piacerti anche
- Casos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSDa EverandCasos de estudio de termodinámica: Solución mediante el uso de ASPENHYSYSValutazione: 4.5 su 5 stelle4.5/5 (7)
- Gases Reales IdealesDocumento13 pagineGases Reales IdealesMelania Garcia ArnezNessuna valutazione finora
- Teoría Del Gas Real: La Relación Exacta Para Gases RealesDa EverandTeoría Del Gas Real: La Relación Exacta Para Gases RealesNessuna valutazione finora
- Gas Real - Wikipedia, La Enciclopedia LibreDocumento22 pagineGas Real - Wikipedia, La Enciclopedia LibreRocio Andrea CasillaNessuna valutazione finora
- Gas RealDocumento17 pagineGas RealEsme PilcoNessuna valutazione finora
- Estudio de Los Gases Ideales y Reales - Lab 1Documento27 pagineEstudio de Los Gases Ideales y Reales - Lab 1Rosario CcahuanticoNessuna valutazione finora
- Segundo Trabajo de TermodinamicaDocumento18 pagineSegundo Trabajo de TermodinamicaAlfonzo Antonio Natera OrtegaNessuna valutazione finora
- IntroIQ Clase 17 y 18Documento27 pagineIntroIQ Clase 17 y 18AR QuímicaNessuna valutazione finora
- Term Odin Á MicaDocumento18 pagineTerm Odin Á MicaJose Pineda RamirezNessuna valutazione finora
- 4.-Modelo Matemático de Redlich-Kwong: R Constante de Los Gases (8.31451 J/mol K)Documento4 pagine4.-Modelo Matemático de Redlich-Kwong: R Constante de Los Gases (8.31451 J/mol K)Katherine JacayNessuna valutazione finora
- Ecuaciones de Estado para Gases RealesDocumento25 pagineEcuaciones de Estado para Gases RealesKEVIN ARNOLDO PEREZ RIVASNessuna valutazione finora
- Estados CorrespondientesDocumento16 pagineEstados CorrespondientesCarlos AlarconNessuna valutazione finora
- Relaciones Termodinamicas Parte 2Documento22 pagineRelaciones Termodinamicas Parte 2Maria caballeroNessuna valutazione finora
- II. Gases Ideales y Sustancia Pura Eq.2Documento54 pagineII. Gases Ideales y Sustancia Pura Eq.2servandoNessuna valutazione finora
- DocumentosDocumento3 pagineDocumentosbetbalon905Nessuna valutazione finora
- Comportamiento de Los GasesDocumento25 pagineComportamiento de Los GasesERIKA LOZADA RUSSELNessuna valutazione finora
- Comportamiento de Los GasesDocumento25 pagineComportamiento de Los GasesElsa Marzana SanizoNessuna valutazione finora
- Ecuaciones de EstadoDocumento10 pagineEcuaciones de EstadoMiguel Angel PalominoNessuna valutazione finora
- Gases Reales y Ecuación de Van Der WaalsDocumento9 pagineGases Reales y Ecuación de Van Der WaalsCamila PachecoNessuna valutazione finora
- Quimica General IDocumento18 pagineQuimica General ISofía Alvarez HernandezNessuna valutazione finora
- Gases Ideales - David M.himmelblauDocumento15 pagineGases Ideales - David M.himmelblauAriel CristóbalNessuna valutazione finora
- Ecuación de Estado para Gases RealesDocumento13 pagineEcuación de Estado para Gases RealesCristian Ccaso MamaniNessuna valutazione finora
- Apuntes QuimicaDocumento13 pagineApuntes QuimicaJAZMIN DIMASNessuna valutazione finora
- GASESDocumento11 pagineGASESFaiberth MoscoteNessuna valutazione finora
- Gases Reales 20-IiDocumento5 pagineGases Reales 20-IiDanny Palma AyalaNessuna valutazione finora
- ExposiciónDocumento5 pagineExposiciónLuis Jose Aguilar MejiaNessuna valutazione finora
- Investigacion Unidad 2. Propiedades de Los Fluidos. Isai ViverosDocumento10 pagineInvestigacion Unidad 2. Propiedades de Los Fluidos. Isai Viverosviveros coloradoNessuna valutazione finora
- Diferencias Entre Gas Real e IdealDocumento7 pagineDiferencias Entre Gas Real e IdealRosmery Orellana Nina100% (2)
- Cap 7 Gases Ideales 19 Mayo 2013 PDFDocumento14 pagineCap 7 Gases Ideales 19 Mayo 2013 PDFKriiztal GodinezNessuna valutazione finora
- Gases Ideales y RealesDocumento6 pagineGases Ideales y RealesjoseNessuna valutazione finora
- Introducción y Conceptos Básicos Ing. Juan Carlos Valdez LoaizaDocumento58 pagineIntroducción y Conceptos Básicos Ing. Juan Carlos Valdez LoaizaSanto 125Nessuna valutazione finora
- Cuestionarios Practica 5Documento8 pagineCuestionarios Practica 5jopisteck01Nessuna valutazione finora
- Clase 5Documento34 pagineClase 5Miguel AngelNessuna valutazione finora
- Comportamiento de Los GasesDocumento25 pagineComportamiento de Los GasesAlfredo G. PradaNessuna valutazione finora
- Gases Ideales VirtuallIIIDocumento11 pagineGases Ideales VirtuallIIIZaida SuniNessuna valutazione finora
- Ley de RaoultDocumento94 pagineLey de RaoultAnonymous idvtpxNessuna valutazione finora
- Asignación Final de Termodinamica IIDocumento8 pagineAsignación Final de Termodinamica IILiz RoncalloNessuna valutazione finora
- Ecuaciones de EstadoDocumento6 pagineEcuaciones de EstadoJüan Prz100% (1)
- Capitulo 4Documento38 pagineCapitulo 4Gabriel MacíasNessuna valutazione finora
- Ecuaciones de Estado para Gases No IdealesDocumento29 pagineEcuaciones de Estado para Gases No IdealesFernando LópezNessuna valutazione finora
- Ecuación de EstadoDocumento127 pagineEcuación de EstadoCarlos CcqNessuna valutazione finora
- Factor de CompresibilidadDocumento5 pagineFactor de CompresibilidadPaola Barbosa100% (7)
- Gases IdealesDocumento12 pagineGases IdealesCinthya MaldonadoNessuna valutazione finora
- Balance de Materia Y Energia PI-111B: Universidad Nacional de Ingenieria Facultad de Ingienieria Quimica Y TextilDocumento38 pagineBalance de Materia Y Energia PI-111B: Universidad Nacional de Ingenieria Facultad de Ingienieria Quimica Y TextilMauricio Norabuena MontesNessuna valutazione finora
- 3 Capacidad Calorifica de Los GasesDocumento30 pagine3 Capacidad Calorifica de Los GasesyesiquisNessuna valutazione finora
- Gases Ideales o PerfectosDocumento31 pagineGases Ideales o PerfectosLuisa M PadovaniNessuna valutazione finora
- Factor de CompresibilidadDocumento6 pagineFactor de CompresibilidadBryanyMariaNessuna valutazione finora
- Modelo Matemático Del Gas RealDocumento4 pagineModelo Matemático Del Gas RealLuna Sherezadee ASNessuna valutazione finora
- Ecuaciones de EstadosDocumento8 pagineEcuaciones de Estadoseduardo fariasNessuna valutazione finora
- Guia de Ejercicios. Corte 1Documento10 pagineGuia de Ejercicios. Corte 1Angely C CastilloNessuna valutazione finora
- Ecuaciones de EstadoDocumento34 pagineEcuaciones de Estadomariacamy07Nessuna valutazione finora
- Gases Ideales y RealesDocumento39 pagineGases Ideales y RealesMaría RicoNessuna valutazione finora
- Problemario Termo IPN Septiembre 2015Documento35 pagineProblemario Termo IPN Septiembre 2015Carol Sán0% (1)
- Ecuaciones de Estado y Factor de CompresibilidadDocumento4 pagineEcuaciones de Estado y Factor de CompresibilidadJesusEduardoPerezPerezNessuna valutazione finora
- Calculo Del Factor Z de Los Gases Por ElDocumento18 pagineCalculo Del Factor Z de Los Gases Por EljeraldoNessuna valutazione finora
- Trabajo de Termo Propiedades ResidualesDocumento25 pagineTrabajo de Termo Propiedades ResidualesLuis Gamero50% (2)
- Ensayo Gas IdealDocumento12 pagineEnsayo Gas Idealjeankarlovelazquez12Nessuna valutazione finora
- Tema Gases RealesDocumento64 pagineTema Gases RealesMaykol AlbercaNessuna valutazione finora
- Gases IdealesDocumento12 pagineGases IdealesRayulss HernandezNessuna valutazione finora
- Informe Fiqui 2 1Documento17 pagineInforme Fiqui 2 1BryanNessuna valutazione finora
- Leccion 15 Principios de Voltametria PDFDocumento5 pagineLeccion 15 Principios de Voltametria PDFAlejandra Adrian TejadaNessuna valutazione finora
- Polisacaridos PDFDocumento17 paginePolisacaridos PDFBrian CeballosNessuna valutazione finora
- Ficha de Orientación y ConsejeríaDocumento4 pagineFicha de Orientación y ConsejeríaAlejandra Adrian Tejada100% (3)
- Diapositivas de SeguridadDocumento30 pagineDiapositivas de SeguridadAlejandra Adrian TejadaNessuna valutazione finora
- Unsa CarDocumento7 pagineUnsa CarAlejandra Adrian TejadaNessuna valutazione finora
- Ejemplo de Práctica de Laboratorio de FísicaDocumento4 pagineEjemplo de Práctica de Laboratorio de FísicaMaría Antón RipollNessuna valutazione finora
- PresentacionDocumento35 paginePresentacionFilipo RvizNessuna valutazione finora
- Informe de Análisis de Tensión IZAJE ACTUALDocumento53 pagineInforme de Análisis de Tensión IZAJE ACTUALLUCCA TONINessuna valutazione finora
- Enfrentando El Modelado de Bioprocesos-Una Revisión de Las Metodologías de ModeladoDocumento19 pagineEnfrentando El Modelado de Bioprocesos-Una Revisión de Las Metodologías de ModeladoLuis VizcaínoNessuna valutazione finora
- Solucionario CienciasDocumento14 pagineSolucionario CienciasJainer RadaNessuna valutazione finora
- Descripcion de Puesto Operador de Planta Rev BDocumento3 pagineDescripcion de Puesto Operador de Planta Rev Brecursos humanosNessuna valutazione finora
- Ejercicios Fluidos en ReposoDocumento3 pagineEjercicios Fluidos en ReposoMateo Ortega PalenciaNessuna valutazione finora
- EXAMEN 2do. QUIMESTRE 20-21 FISICADocumento2 pagineEXAMEN 2do. QUIMESTRE 20-21 FISICAOskar HumananteNessuna valutazione finora
- Efecto de La Temperatura Sobre La Rapidez de La Reacción.....Documento9 pagineEfecto de La Temperatura Sobre La Rapidez de La Reacción.....saul corona100% (1)
- EutrofizacionDocumento16 pagineEutrofizacionElva DanaeNessuna valutazione finora
- MSDS Aceite Hydra 68 LubraxDocumento9 pagineMSDS Aceite Hydra 68 LubraxNaJo CasBarNessuna valutazione finora
- Mezclas AlcalinasDocumento17 pagineMezclas AlcalinasAlexis John Soncco HanccoNessuna valutazione finora
- Análisis CacaoDocumento21 pagineAnálisis CacaoPaulandrea MoraNessuna valutazione finora
- Ficometa Labo 2Documento10 pagineFicometa Labo 2Carlos Rivas MinayaNessuna valutazione finora
- Cuándo Se Formó El Sistema SolarDocumento4 pagineCuándo Se Formó El Sistema SolarHanna Elías CamposNessuna valutazione finora
- Valoracion Redox FinalDocumento29 pagineValoracion Redox FinalebertNessuna valutazione finora
- Pia de Procesos de ManufacturaDocumento13 paginePia de Procesos de ManufacturaEdith SaldañaNessuna valutazione finora
- Informe Determinación de Colorante en Bebidas - FinalDocumento8 pagineInforme Determinación de Colorante en Bebidas - FinalJORGE LEONARDO FRASSER QUIÑONES0% (1)
- SUMITEN780SDocumento42 pagineSUMITEN780SLuis Chiara LoayzaNessuna valutazione finora
- Guia 3 Septimo 2 Periodo 20214Documento5 pagineGuia 3 Septimo 2 Periodo 20214Luis CabariqueNessuna valutazione finora
- Voltage Inducido en Una Espira GiratoriaDocumento16 pagineVoltage Inducido en Una Espira Giratoriaandres atencioNessuna valutazione finora
- Cambios de Fase Resuelta. WordDocumento4 pagineCambios de Fase Resuelta. WordJuan Almonacid GallardoNessuna valutazione finora
- Produccion y Aplicaciones Del BiogásDocumento12 pagineProduccion y Aplicaciones Del BiogásdiegoNessuna valutazione finora
- Diagrama OleorresinasDocumento69 pagineDiagrama OleorresinaskenethNessuna valutazione finora
- Guia de Laboratorio de Fisica 2 2018-2 PDFDocumento75 pagineGuia de Laboratorio de Fisica 2 2018-2 PDFAlexis Mariano R SantillanNessuna valutazione finora
- PH Problemas Fa Tima Condori LazoDocumento11 paginePH Problemas Fa Tima Condori LazoCristell LazoNessuna valutazione finora
- Lubricante Golden Bear RematerisadoDocumento11 pagineLubricante Golden Bear RematerisadoMarlon MuzoNessuna valutazione finora
- Charla Seguridad Reactivos MerckDocumento105 pagineCharla Seguridad Reactivos MerckJuan M MasteryNessuna valutazione finora
- 7 Unidad VII. QMA-103Documento10 pagine7 Unidad VII. QMA-103Omar SantosNessuna valutazione finora
- Informe 2Documento7 pagineInforme 2David Alejandro RamirezNessuna valutazione finora