Potrebbero piacerti anche
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesDa EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNessuna valutazione finora
- Synthesis of N-Chloroquinolines and N-Ethynylquinolines (nZ2, 4, 8) : Homo and Heterocoupling Reactions.Documento10 pagineSynthesis of N-Chloroquinolines and N-Ethynylquinolines (nZ2, 4, 8) : Homo and Heterocoupling Reactions.kawtherahmedNessuna valutazione finora
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesDa EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesNessuna valutazione finora
- Etard ReactionDocumento5 pagineEtard Reactionp3pumNessuna valutazione finora
- Asian J. Org. Chem. 2015, 4, 28 - 32 PDFDocumento5 pagineAsian J. Org. Chem. 2015, 4, 28 - 32 PDFSulagna DasNessuna valutazione finora
- A New Method For The Synthesis of Aliphatic Nitro Compounds1, 2Documento5 pagineA New Method For The Synthesis of Aliphatic Nitro Compounds1, 2banjo01Nessuna valutazione finora
- 1,5-Dipolar Cyclizations: 1. LntroducfionDocumento51 pagine1,5-Dipolar Cyclizations: 1. LntroducfionRikta SahaNessuna valutazione finora
- Nitrosation of Peptide BondDocumento7 pagineNitrosation of Peptide BondEugenia CebanNessuna valutazione finora
- Lewis Acid Promoted Reactions of N - (Chlorides. Ring-Size Effects in Competitive Intramolecular Acylation of Phenyl and Cyclopropyl SubstituentsDocumento3 pagineLewis Acid Promoted Reactions of N - (Chlorides. Ring-Size Effects in Competitive Intramolecular Acylation of Phenyl and Cyclopropyl SubstituentsthamtusieuquayNessuna valutazione finora
- Thiophenol SynthesisDocumento6 pagineThiophenol SynthesisNirav ShahNessuna valutazione finora
- Expedient Access To Unsymmetrical Diarylindolylmethanes Through Palladium-Catalyzed Domino Electrophilic Cyclization Extended Conjugate Addition ApproachDocumento4 pagineExpedient Access To Unsymmetrical Diarylindolylmethanes Through Palladium-Catalyzed Domino Electrophilic Cyclization Extended Conjugate Addition ApproachMR.ZUHAIB MughalNessuna valutazione finora
- Decompositions of Di-t-Alkyl Peroxides. I. Kinetics: Frederick F. EDocumento7 pagineDecompositions of Di-t-Alkyl Peroxides. I. Kinetics: Frederick F. Emartinml_1191Nessuna valutazione finora
- Synthesis of Levulinic Acid-Glycerol Ketal-Ester Oligomers and Structural Characterization Using NMR SpectrosDocumento7 pagineSynthesis of Levulinic Acid-Glycerol Ketal-Ester Oligomers and Structural Characterization Using NMR SpectrosLucas de MeloNessuna valutazione finora
- Synthesis and Evaluation of Some Variants of The Nefkens' ReagentDocumento3 pagineSynthesis and Evaluation of Some Variants of The Nefkens' Reagentlost6taNessuna valutazione finora
- New Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsDocumento6 pagineNew Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsChamula K MasNessuna valutazione finora
- Chem. Eur. J. 2011, 17, 10208 - 10212Documento5 pagineChem. Eur. J. 2011, 17, 10208 - 10212SBNessuna valutazione finora
- 1 s2.0 S0926860X08000963 MainDocumento9 pagine1 s2.0 S0926860X08000963 Mainpetru apopeiNessuna valutazione finora
- Kinetics of The Ring-Opening Polymerization Of, - Lactide Using Zinc (II) Octoate As CatalystDocumento9 pagineKinetics of The Ring-Opening Polymerization Of, - Lactide Using Zinc (II) Octoate As CatalystGift GibbNessuna valutazione finora
- Tetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi YuDocumento4 pagineTetrahedron Letters: Shan Wang, Jian Wang, Rui Guo, Gao Wang, Shan-Yong Chen, Xiao-Qi Yumarkbronson2009Nessuna valutazione finora
- The Role and Reactions of Nitroxyl Radicals in Hindered Piperidine Light StabilisationDocumento8 pagineThe Role and Reactions of Nitroxyl Radicals in Hindered Piperidine Light StabilisationStacey DongNessuna valutazione finora
- Paper: Microwave Methods For The Synthesis of Paddlewheel Diruthenium Compounds With N, N-Donor LigandsDocumento6 paginePaper: Microwave Methods For The Synthesis of Paddlewheel Diruthenium Compounds With N, N-Donor LigandshttmaiNessuna valutazione finora
- Kinetics of Iodination of Acetone Catalyzed by HCL and H2so4 A Colorimetric Investigation of Relative StrengthDocumento2 pagineKinetics of Iodination of Acetone Catalyzed by HCL and H2so4 A Colorimetric Investigation of Relative StrengthHansel VereitelnNessuna valutazione finora
- P 19900000945Documento10 pagineP 19900000945Yi-Yan TsaoNessuna valutazione finora
- PD CatDocumento7 paginePD CatKiss LeviNessuna valutazione finora
- Review of Blocked and Deblocked IsocyanateDocumento8 pagineReview of Blocked and Deblocked IsocyanateAdlyLubisNessuna valutazione finora
- R.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeDocumento5 pagineR.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeGummyColaNessuna valutazione finora
- An Experiment For Undergraduate Advanced Inorganic Chemistry StudentsDocumento19 pagineAn Experiment For Undergraduate Advanced Inorganic Chemistry StudentsKiki AimaNessuna valutazione finora
- A Practical Synthesis of (-) - Kainic AcidDocumento11 pagineA Practical Synthesis of (-) - Kainic AcidNikhil SarpateNessuna valutazione finora
- Com 97 8051Documento5 pagineCom 97 8051bruna_0410Nessuna valutazione finora
- 1971 R K Kulkarni E G Moore A F Hegyeli - Fred Leonard (1971) - Biodegradable Poly (Lactic Acid) Polymers 5 (3) 169-181 1971Documento13 pagine1971 R K Kulkarni E G Moore A F Hegyeli - Fred Leonard (1971) - Biodegradable Poly (Lactic Acid) Polymers 5 (3) 169-181 1971Julio ArruaNessuna valutazione finora
- Titanium FractionDocumento9 pagineTitanium FractionAhmadNessuna valutazione finora
- Tetrahedron Letters Volume Issue 2016 (Doi 10.1016/j.tetlet.2016.04.112) Nagarajan, Rajendran Jayashankaran, Jayadevan Emmanuvel, Lourd - Transition Metal-Free Steric Controlled One - Pot SynthesDocumento13 pagineTetrahedron Letters Volume Issue 2016 (Doi 10.1016/j.tetlet.2016.04.112) Nagarajan, Rajendran Jayashankaran, Jayadevan Emmanuvel, Lourd - Transition Metal-Free Steric Controlled One - Pot SynthesJayash RulzNessuna valutazione finora
- Mo JAEDocumento4 pagineMo JAEThanhThao TranNessuna valutazione finora
- Angew. Chem. Int. Ed. 2011, 50, 6167 - 6170Documento4 pagineAngew. Chem. Int. Ed. 2011, 50, 6167 - 6170NoimurNessuna valutazione finora
- Preparation of A Polysulfide: RubberDocumento1 paginaPreparation of A Polysulfide: RubberRicky EstepaNessuna valutazione finora
- A Review of Organotin Compounds Chemistry and ApplDocumento9 pagineA Review of Organotin Compounds Chemistry and ApplJuan C HernandezNessuna valutazione finora
- Datta 2012Documento4 pagineDatta 2012SakhaviTVNessuna valutazione finora
- Aryl GrignardDocumento4 pagineAryl GrignardRoss LewinNessuna valutazione finora
- Glyconanoparticles Activity 3Documento3 pagineGlyconanoparticles Activity 3ЛуизАпазаТ.Nessuna valutazione finora
- Kim2002 - PolikondensasiDocumento6 pagineKim2002 - PolikondensasiMathilda PasaribuNessuna valutazione finora
- 檔案下載Documento6 pagine檔案下載Rosalina DjatmikaNessuna valutazione finora
- The Room Temperature Polymerization of Propylene OxideDocumento5 pagineThe Room Temperature Polymerization of Propylene OxidecesarmachucaNessuna valutazione finora
- Synthesis, Structure and Chemical Transformations of 4-AminobenzaldehydeDocumento5 pagineSynthesis, Structure and Chemical Transformations of 4-AminobenzaldehydeBrem BalazsNessuna valutazione finora
- FeCl3-Catalyzed Synthesis of 2-Methyl-4-Substituted-1,2,3,4-Tetrahydroquinoline DerivativesDocumento4 pagineFeCl3-Catalyzed Synthesis of 2-Methyl-4-Substituted-1,2,3,4-Tetrahydroquinoline DerivativesRajesh TammanaNessuna valutazione finora
- Asian J. Org. Chem. 2021,10, 2152-2156Documento5 pagineAsian J. Org. Chem. 2021,10, 2152-2156NoimurNessuna valutazione finora
- Isolation, Biological Activities and Synthesis of Indoloquinoline Alkaloids: Cryptole-Pine, Isocryptolepine and NeocryptolepineDocumento22 pagineIsolation, Biological Activities and Synthesis of Indoloquinoline Alkaloids: Cryptole-Pine, Isocryptolepine and NeocryptolepineDayse_sbNessuna valutazione finora
- Tetrahedron 64 (2008) 219e233 - RosyDocumento15 pagineTetrahedron 64 (2008) 219e233 - RosyRamdas BorhadeNessuna valutazione finora
- Exp't 13: Phase-Transfer-Catalyzed Alkylation of Diethyl MalonateDocumento5 pagineExp't 13: Phase-Transfer-Catalyzed Alkylation of Diethyl MalonatelovehopeNessuna valutazione finora
- ReductionDocumento2 pagineReductionCatenaneNessuna valutazione finora
- tmp2AEF TMPDocumento6 paginetmp2AEF TMPFrontiersNessuna valutazione finora
- Synthesis, Characterization and Application of O, O-Diethyl Acrylamide PhosphateDocumento5 pagineSynthesis, Characterization and Application of O, O-Diethyl Acrylamide Phosphateandi tiaNessuna valutazione finora
- Chiral Sulphonated Phosphines. Part VII. Catalytic Transfer-Hydrogenation of Unsaturated Substrates With Formates in The Presence of Water Soluble Complexes of RhodaDocumento4 pagineChiral Sulphonated Phosphines. Part VII. Catalytic Transfer-Hydrogenation of Unsaturated Substrates With Formates in The Presence of Water Soluble Complexes of RhodappopgodNessuna valutazione finora
- MR14 5 0510Documento7 pagineMR14 5 0510rusheekesh3497Nessuna valutazione finora
- (Grupo 3) Wang-2021-Ligandcontrolled-tunable-coppercataDocumento8 pagine(Grupo 3) Wang-2021-Ligandcontrolled-tunable-coppercataVANESSA RODRIGUEZ CEBALLOSNessuna valutazione finora
- Strategies Toward The Design of Energetic Ionic Liquids: Nitro-And Nitrile-Substituted N, N - Dialkylimidazolium SaltswDocumento10 pagineStrategies Toward The Design of Energetic Ionic Liquids: Nitro-And Nitrile-Substituted N, N - Dialkylimidazolium SaltswZhan FangNessuna valutazione finora
- 12 - Xu - JMolCatA - Dihydrochalcones Via The AlCl3 CynamoylDocumento9 pagine12 - Xu - JMolCatA - Dihydrochalcones Via The AlCl3 Cynamoyllenggah purwandariNessuna valutazione finora
- Standard Molar Enthalpies of Formation of Crystalline Stereoisomers of Aldono-1,4-LactonesDocumento6 pagineStandard Molar Enthalpies of Formation of Crystalline Stereoisomers of Aldono-1,4-LactonesManuNessuna valutazione finora
- Luminescence Probe Studies of Nafion PolyelectrolytesDocumento5 pagineLuminescence Probe Studies of Nafion PolyelectrolytesLuis AlvarezNessuna valutazione finora
- Binol On SilicaDocumento8 pagineBinol On SilicaMinal ButalaNessuna valutazione finora
- Sodium Nitrite Catalyzed Aerobic Oxidative Deoximation Under Mild ConditionsDocumento4 pagineSodium Nitrite Catalyzed Aerobic Oxidative Deoximation Under Mild ConditionsCláudio SerafimNessuna valutazione finora
- MSDS Polyamine - Bluwat ChemicalsDocumento4 pagineMSDS Polyamine - Bluwat ChemicalsAiza CabolesNessuna valutazione finora
- Sun Life ProspectusDocumento39 pagineSun Life ProspectusAiza CabolesNessuna valutazione finora
- Iw 8 BenDocumento7 pagineIw 8 BenAdien GumilangNessuna valutazione finora
- Base ConversionDocumento2 pagineBase ConversionAiza CabolesNessuna valutazione finora
- Delta To Wye and V.VDocumento4 pagineDelta To Wye and V.VAiza CabolesNessuna valutazione finora
- Crayon ReportDocumento4 pagineCrayon ReportAiza CabolesNessuna valutazione finora
- Critique PaperDocumento1 paginaCritique PaperAiza CabolesNessuna valutazione finora
- Complete List AutoCAD AliasesDocumento11 pagineComplete List AutoCAD AliasesAiza CabolesNessuna valutazione finora
- PolyphenolsDocumento12 paginePolyphenolsiyanarak8475Nessuna valutazione finora
- Base Conversion PDFDocumento2 pagineBase Conversion PDFAiza CabolesNessuna valutazione finora
- Sample Calculations For Surface TensionDocumento6 pagineSample Calculations For Surface TensionAiza CabolesNessuna valutazione finora
- How To Do Lattice MultiplicationDocumento3 pagineHow To Do Lattice MultiplicationAiza CabolesNessuna valutazione finora
- BSChE FlowchartDocumento3 pagineBSChE FlowchartAiza CabolesNessuna valutazione finora
- US Sieve SeriesDocumento4 pagineUS Sieve SeriesAiza CabolesNessuna valutazione finora
- DR Perla D Santos OcampoDocumento2 pagineDR Perla D Santos OcampoAiza CabolesNessuna valutazione finora
- How To Calculate TorqueDocumento6 pagineHow To Calculate TorqueAiza Caboles100% (1)
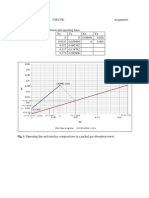
- Table 1. Data For The Equilibrium and Operating Lines.: Caboles, Ellaiza Marie G. Che153E Assignment 2011-45827Documento1 paginaTable 1. Data For The Equilibrium and Operating Lines.: Caboles, Ellaiza Marie G. Che153E Assignment 2011-45827Aiza CabolesNessuna valutazione finora
- ChE 192 Handout 6a (Storage Tank)Documento4 pagineChE 192 Handout 6a (Storage Tank)Aiza CabolesNessuna valutazione finora
- How To Do Lattice MultiplicationDocumento3 pagineHow To Do Lattice MultiplicationAiza CabolesNessuna valutazione finora
- UOP ParaxyleneDocumento2 pagineUOP ParaxyleneAiza CabolesNessuna valutazione finora
- Oxygen Bomb CalorimeterDocumento5 pagineOxygen Bomb CalorimeterAiza CabolesNessuna valutazione finora
- STHE Specifications SheetDocumento8 pagineSTHE Specifications SheetAiza CabolesNessuna valutazione finora
- Eubp Factsfigures Bioplastics 2013Documento6 pagineEubp Factsfigures Bioplastics 2013Aiza CabolesNessuna valutazione finora
- Che 154 Lecture 3b Screening EquipmentDocumento34 pagineChe 154 Lecture 3b Screening EquipmentAiza CabolesNessuna valutazione finora
- Experimental Determination of The Joule Thomson Coefficient of Nitrogen Carbon Dioxide and OxygenDocumento5 pagineExperimental Determination of The Joule Thomson Coefficient of Nitrogen Carbon Dioxide and OxygenAiza CabolesNessuna valutazione finora
- Joule-Thomson Coefficient: Experiment T9 Chemistry 114Documento4 pagineJoule-Thomson Coefficient: Experiment T9 Chemistry 114truffeloveNessuna valutazione finora
- Eubp Factsfigures Bioplastics 2013Documento6 pagineEubp Factsfigures Bioplastics 2013Aiza CabolesNessuna valutazione finora
- Table 1. Values Computed For The Multicomponent Absorption DiagramDocumento1 paginaTable 1. Values Computed For The Multicomponent Absorption DiagramAiza CabolesNessuna valutazione finora
- Forest Ecology Vocab TermsDocumento2 pagineForest Ecology Vocab TermsKapetan BarbarosaNessuna valutazione finora
- Safety Data Sheet: 1 IdentificationDocumento11 pagineSafety Data Sheet: 1 IdentificationKara WhiteNessuna valutazione finora
- USP Monographs - Monobasic Sodium PhosphateDocumento2 pagineUSP Monographs - Monobasic Sodium PhosphateGanesh KashinathNessuna valutazione finora
- 10 Science Notes 03 Metals and Non Metals 1Documento9 pagine10 Science Notes 03 Metals and Non Metals 1varunNessuna valutazione finora
- B.pharm Ruhs SyllabusDocumento41 pagineB.pharm Ruhs Syllabusgopi01857% (7)
- CHAPTER 6 ElctrochemistryDocumento8 pagineCHAPTER 6 ElctrochemistryMohd Nazri Mat JaridNessuna valutazione finora
- Mercuric Chloride: Safety Data SheetDocumento10 pagineMercuric Chloride: Safety Data SheetWeare1_busyNessuna valutazione finora
- Coating SDocumento51 pagineCoating SBalaji GuruNessuna valutazione finora
- Latihan PDPR Kimia SPM 2021 Pada 21 AprilDocumento8 pagineLatihan PDPR Kimia SPM 2021 Pada 21 Aprilexo kaihunNessuna valutazione finora
- Al Ms App Catalog Index PDFDocumento108 pagineAl Ms App Catalog Index PDFapc108Nessuna valutazione finora
- Amber AnalysisDocumento34 pagineAmber AnalysisKonstantinos Angelidakis100% (1)
- Manufacturing Fine TiO2 Particles by Chloride ProcessDocumento2 pagineManufacturing Fine TiO2 Particles by Chloride ProcessDiahAyuSafitriNessuna valutazione finora
- Chem 2 Chemistry in Your World 2nd Edition Hogg Solutions ManualDocumento38 pagineChem 2 Chemistry in Your World 2nd Edition Hogg Solutions Manualgarrywolfelsjftl100% (14)
- Ndoro Corrected DissertationDocumento78 pagineNdoro Corrected DissertationOnesime Muteba100% (1)
- IntroductionDocumento29 pagineIntroductionanamendoza1868Nessuna valutazione finora
- Mineral WebquestDocumento3 pagineMineral Webquestapi-268569185Nessuna valutazione finora
- CBSE 10th Science Sample Paper 1Documento5 pagineCBSE 10th Science Sample Paper 1Aditya AcharyaNessuna valutazione finora
- Conscise Biochemistry MCQDocumento27 pagineConscise Biochemistry MCQRavi Bhatnagar100% (1)
- Jurnal Cetrimide 2Documento5 pagineJurnal Cetrimide 2TogiNessuna valutazione finora
- Agus2018 Article CompositionOfUnfermentedUnroasDocumento9 pagineAgus2018 Article CompositionOfUnfermentedUnroasnubnubNessuna valutazione finora
- Lab Report Experiment 3Documento4 pagineLab Report Experiment 3Juan TlilayatziNessuna valutazione finora
- TanninsDocumento20 pagineTanninsnotapernota101100% (1)
- Synthesis of Aspirin DataDocumento3 pagineSynthesis of Aspirin DataAnonymous orNHXM0f0Nessuna valutazione finora
- NtrenmDocumento16 pagineNtrenmNicoleta IorgaNessuna valutazione finora
- Science Year 7 Summer Acids and AlkalisDocumento1 paginaScience Year 7 Summer Acids and AlkalisMfanafuthiNessuna valutazione finora
- Dispensette: The Standard in Bottletop Dispensing For A Half CenturyDocumento16 pagineDispensette: The Standard in Bottletop Dispensing For A Half CenturyDiana CastilloNessuna valutazione finora
- E1mdlcsm Rev1122Documento15 pagineE1mdlcsm Rev1122Pkk Siam Rayong co-saleNessuna valutazione finora
- Hydrogenation of Nitrobenzene To P-Aminophenol in A Four-Phase Reactor Reaction Kinetics and Mass Transfer EffectsDocumento6 pagineHydrogenation of Nitrobenzene To P-Aminophenol in A Four-Phase Reactor Reaction Kinetics and Mass Transfer EffectsHeylenLoperaNessuna valutazione finora
- Pitiagi 22 P Abs 045Documento7 paginePitiagi 22 P Abs 045Amira SiregarNessuna valutazione finora
- CBSE Class 12 Chemistry Practice Paper 2017Documento5 pagineCBSE Class 12 Chemistry Practice Paper 2017rishi017Nessuna valutazione finora
- ChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindDa EverandChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindNessuna valutazione finora
- Hero Found: The Greatest POW Escape of the Vietnam WarDa EverandHero Found: The Greatest POW Escape of the Vietnam WarValutazione: 4 su 5 stelle4/5 (19)
- Sully: The Untold Story Behind the Miracle on the HudsonDa EverandSully: The Untold Story Behind the Miracle on the HudsonValutazione: 4 su 5 stelle4/5 (103)
- The End of Craving: Recovering the Lost Wisdom of Eating WellDa EverandThe End of Craving: Recovering the Lost Wisdom of Eating WellValutazione: 4.5 su 5 stelle4.5/5 (82)
- The Fabric of Civilization: How Textiles Made the WorldDa EverandThe Fabric of Civilization: How Textiles Made the WorldValutazione: 4.5 su 5 stelle4.5/5 (58)
- Fire on the Horizon: The Untold Story of the Gulf Oil DisasterDa EverandFire on the Horizon: The Untold Story of the Gulf Oil DisasterNessuna valutazione finora
- How to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerDa EverandHow to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerValutazione: 4.5 su 5 stelle4.5/5 (122)
- Highest Duty: My Search for What Really MattersDa EverandHighest Duty: My Search for What Really MattersNessuna valutazione finora
- Reality+: Virtual Worlds and the Problems of PhilosophyDa EverandReality+: Virtual Worlds and the Problems of PhilosophyValutazione: 4 su 5 stelle4/5 (24)
- System Error: Where Big Tech Went Wrong and How We Can RebootDa EverandSystem Error: Where Big Tech Went Wrong and How We Can RebootNessuna valutazione finora
- The Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaDa EverandThe Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaNessuna valutazione finora
- Faster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestDa EverandFaster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestValutazione: 4 su 5 stelle4/5 (28)
- Dirt to Soil: One Family’s Journey into Regenerative AgricultureDa EverandDirt to Soil: One Family’s Journey into Regenerative AgricultureValutazione: 5 su 5 stelle5/5 (125)
- The Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyDa EverandThe Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyNessuna valutazione finora
- Pale Blue Dot: A Vision of the Human Future in SpaceDa EverandPale Blue Dot: A Vision of the Human Future in SpaceValutazione: 4.5 su 5 stelle4.5/5 (588)
- A Place of My Own: The Architecture of DaydreamsDa EverandA Place of My Own: The Architecture of DaydreamsValutazione: 4 su 5 stelle4/5 (242)
- How to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerDa EverandHow to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerValutazione: 4.5 su 5 stelle4.5/5 (54)
- Broken Money: Why Our Financial System is Failing Us and How We Can Make it BetterDa EverandBroken Money: Why Our Financial System is Failing Us and How We Can Make it BetterValutazione: 5 su 5 stelle5/5 (3)
- Packing for Mars: The Curious Science of Life in the VoidDa EverandPacking for Mars: The Curious Science of Life in the VoidValutazione: 4 su 5 stelle4/5 (1396)
- The Future of Geography: How the Competition in Space Will Change Our WorldDa EverandThe Future of Geography: How the Competition in Space Will Change Our WorldValutazione: 4 su 5 stelle4/5 (5)
- The Assassination Complex: Inside the Government's Secret Drone Warfare ProgramDa EverandThe Assassination Complex: Inside the Government's Secret Drone Warfare ProgramValutazione: 4 su 5 stelle4/5 (55)
- The Book of the Moon: A Guide to Our Closest NeighborDa EverandThe Book of the Moon: A Guide to Our Closest NeighborValutazione: 4.5 su 5 stelle4.5/5 (11)
- The Things We Make: The Unknown History of Invention from Cathedrals to Soda CansDa EverandThe Things We Make: The Unknown History of Invention from Cathedrals to Soda CansNessuna valutazione finora
- The Weather Machine: A Journey Inside the ForecastDa EverandThe Weather Machine: A Journey Inside the ForecastValutazione: 3.5 su 5 stelle3.5/5 (31)
- The Knowledge: How to Rebuild Our World from ScratchDa EverandThe Knowledge: How to Rebuild Our World from ScratchValutazione: 3.5 su 5 stelle3.5/5 (133)
- Permaculture for the Rest of Us: Abundant Living on Less than an AcreDa EverandPermaculture for the Rest of Us: Abundant Living on Less than an AcreValutazione: 4.5 su 5 stelle4.5/5 (33)
- Transformed: Moving to the Product Operating ModelDa EverandTransformed: Moving to the Product Operating ModelValutazione: 4 su 5 stelle4/5 (1)