Potrebbero piacerti anche
- Monoclonal Antibody and Immunosensor Technology: <b>The production and application of rodent and human monoclonal antibodies</b>Da EverandMonoclonal Antibody and Immunosensor Technology: <b>The production and application of rodent and human monoclonal antibodies</b>Nessuna valutazione finora
- AP Biology Lab Six A and B: DNA Fingerprinting and Bacterial TransformationDocumento9 pagineAP Biology Lab Six A and B: DNA Fingerprinting and Bacterial TransformationCoolAsianDude81% (26)
- New Approaches in Cell Biology: Proceedings of a Symposium Held At Imperial College, London, July 1958Da EverandNew Approaches in Cell Biology: Proceedings of a Symposium Held At Imperial College, London, July 1958Nessuna valutazione finora
- Lab 3 BMB 442 FinishedDocumento13 pagineLab 3 BMB 442 Finishedapi-260887014Nessuna valutazione finora
- Research in ProtozoologyDa EverandResearch in ProtozoologyTze-Tuan ChenValutazione: 5 su 5 stelle5/5 (1)
- Transformation BiotechnologyDocumento3 pagineTransformation BiotechnologyAseem GuptaNessuna valutazione finora
- Mechanisms of Eukaryotic DNA RecombinationDa EverandMechanisms of Eukaryotic DNA RecombinationMax E GottesmanNessuna valutazione finora
- Vibrio Fischeri, Which Naturally Lives in The Light Organ of The Fish Monocentris Japonicas. Together, TheseDocumento3 pagineVibrio Fischeri, Which Naturally Lives in The Light Organ of The Fish Monocentris Japonicas. Together, TheseLathia WhitakerNessuna valutazione finora
- Biotechnology: A Laboratory CourseDa EverandBiotechnology: A Laboratory CourseValutazione: 5 su 5 stelle5/5 (1)
- Gene TransformationDocumento13 pagineGene TransformationFelix Ivander SalimNessuna valutazione finora
- APBIO - pGLO Lab ReportDocumento5 pagineAPBIO - pGLO Lab Reportekulps100% (4)
- AP Biology Lab 6 TransformationDocumento3 pagineAP Biology Lab 6 TransformationLogan GreenNessuna valutazione finora
- Practical Report - BiotechnologyDocumento23 paginePractical Report - Biotechnologylightning proNessuna valutazione finora
- Biology - Lab 4 (The Bacterial Transformation Lab)Documento4 pagineBiology - Lab 4 (The Bacterial Transformation Lab)Evans LoveNessuna valutazione finora
- Bacterial Transformation: The Lab Protocol Can Be Conceptualized As Four Major Steps. Pre-IncubationDocumento4 pagineBacterial Transformation: The Lab Protocol Can Be Conceptualized As Four Major Steps. Pre-IncubationGUIDO ERNESTO VILLOTA CALVACHINessuna valutazione finora
- Bacterial Transformation Lab (6a)Documento7 pagineBacterial Transformation Lab (6a)Chris PriceNessuna valutazione finora
- Competent Cells and TransformationDocumento22 pagineCompetent Cells and TransformationCarinaJongLeeNessuna valutazione finora
- Gene Transfer FinishedDocumento5 pagineGene Transfer FinishedTheoNessuna valutazione finora
- WINSEM2023-24 BBIT302P LO VL2023240505753 2024-02-24 Reference-Material-IDocumento3 pagineWINSEM2023-24 BBIT302P LO VL2023240505753 2024-02-24 Reference-Material-IVibhawari Ramakrishna Wusirika 21BBT0386Nessuna valutazione finora
- Gene TransformationDocumento6 pagineGene TransformationFelix Ivander SalimNessuna valutazione finora
- AP Lab #6: pGLO Transformation LabDocumento4 pagineAP Lab #6: pGLO Transformation Labpointweb85% (20)
- Transformationandtransfectionfinal 180609201420 PDFDocumento35 pagineTransformationandtransfectionfinal 180609201420 PDFDarshan MarjadiNessuna valutazione finora
- Ap Lab 6Documento3 pagineAp Lab 6api-224488631Nessuna valutazione finora
- Bacterial Transformation 2 FinalDocumento16 pagineBacterial Transformation 2 FinalRianAidil100% (1)
- Acb Transformation Efficiency PracticalDocumento10 pagineAcb Transformation Efficiency PracticalGanesh KumarNessuna valutazione finora
- Pgal Transformation Complete Lab Revised-1Documento21 paginePgal Transformation Complete Lab Revised-1davidNessuna valutazione finora
- Gene Expression Lab LogDocumento7 pagineGene Expression Lab LogjenniferNessuna valutazione finora
- Research Papers On Bacterial TransformationDocumento4 pagineResearch Papers On Bacterial TransformationlihbcfvkgNessuna valutazione finora
- Vectores y PlasmidosDocumento7 pagineVectores y PlasmidosSarita UrbanoNessuna valutazione finora
- Lab 3 Lab ManualDocumento3 pagineLab 3 Lab Manualmakabigail7Nessuna valutazione finora
- P GLOBacterial Transformation ProtocolDocumento8 pagineP GLOBacterial Transformation Protocoljexmix0608Nessuna valutazione finora
- mcb130 Sci PaperDocumento9 paginemcb130 Sci PaperShannen SenaNessuna valutazione finora
- Pglo Transformation Lab Report - Kierra LeonardDocumento9 paginePglo Transformation Lab Report - Kierra Leonardapi-508539249Nessuna valutazione finora
- AP Biology Transformation Lab ReportDocumento3 pagineAP Biology Transformation Lab Reportdstaples7100% (17)
- Recent Research Papers On Molecular BiologyDocumento6 pagineRecent Research Papers On Molecular Biologyozbvtcvkg100% (1)
- BFP To GFPDocumento11 pagineBFP To GFPKeri Gobin SamarooNessuna valutazione finora
- Second Version Research PaperDocumento6 pagineSecond Version Research PaperSwetha ThiagarajanNessuna valutazione finora
- Bacterial Transformation Lab ReportDocumento11 pagineBacterial Transformation Lab ReportRichie JustinNessuna valutazione finora
- Gram Positive and Gram Negative Bacteria Differ inDocumento12 pagineGram Positive and Gram Negative Bacteria Differ inWidya Noor HalizaNessuna valutazione finora
- GENETICSDocumento26 pagineGENETICSANIRBAN PALNessuna valutazione finora
- Ligation and TransformationDocumento6 pagineLigation and TransformationCarina JLNessuna valutazione finora
- Sel KompetenDocumento12 pagineSel KompetenEnung Warsita DahlanNessuna valutazione finora
- Budding Yeast Genetics ExperimentDocumento15 pagineBudding Yeast Genetics Experimentapi-720097976Nessuna valutazione finora
- Light The: Regulation of Cell Cycle inDocumento7 pagineLight The: Regulation of Cell Cycle inTeresa MataNessuna valutazione finora
- Student Manual pGLO Transformation: Lesson 1 Introduction To TransformationDocumento26 pagineStudent Manual pGLO Transformation: Lesson 1 Introduction To TransformationanamqadirNessuna valutazione finora
- Student Manual pGLO Transformation: Lesson 1 Introduction To TransformationDocumento21 pagineStudent Manual pGLO Transformation: Lesson 1 Introduction To TransformationHassan GarridoNessuna valutazione finora
- Ashbya Gossypii Under Osmotic StressDocumento6 pagineAshbya Gossypii Under Osmotic Stresscharchar911Nessuna valutazione finora
- CDNA Lab Report - Docx2Documento4 pagineCDNA Lab Report - Docx2cxs5278100% (1)
- Lab Manual Molecular BiologyDocumento19 pagineLab Manual Molecular BiologyLockerLingNessuna valutazione finora
- Chap 12Documento86 pagineChap 12chemptnkNessuna valutazione finora
- 4 - Guided Learning Questions - Chapter 3.1-3.3Documento5 pagine4 - Guided Learning Questions - Chapter 3.1-3.3ashleighewilkinsonNessuna valutazione finora
- Problem Set 51-100Documento11 pagineProblem Set 51-100Penelope LeizonNessuna valutazione finora
- Vectores de TransferenciaDocumento7 pagineVectores de TransferenciaAlex Guijarro PadillaNessuna valutazione finora
- Cloning A DNA Segment From Bacteriophage LambdaDocumento13 pagineCloning A DNA Segment From Bacteriophage LambdaBilypede55Nessuna valutazione finora
- DNA RecombinationnewDocumento34 pagineDNA Recombinationnewsecretary.kimeniahNessuna valutazione finora
- Competent Cell Prep, Transformation Protocol (LAB 1 & 2)Documento3 pagineCompetent Cell Prep, Transformation Protocol (LAB 1 & 2)Mehul darakNessuna valutazione finora
- Formal Lab Report 1Documento7 pagineFormal Lab Report 1the_real_wasabiNessuna valutazione finora
- How Does Plasmid DNA Penetrate Cell Membranes in Artificial Transformation Process of Escherichia ColiDocumento13 pagineHow Does Plasmid DNA Penetrate Cell Membranes in Artificial Transformation Process of Escherichia Colibcn574835Nessuna valutazione finora
- 12 Yeast Transformation 2013 1Documento8 pagine12 Yeast Transformation 2013 1ingeneria biotecnologicaNessuna valutazione finora
- ProtocolDocumento2 pagineProtocolShruti PillaiNessuna valutazione finora
- Aquarius. As A Modern Work, The Typical Audience Member Is Separated by at Least A GenerationDocumento5 pagineAquarius. As A Modern Work, The Typical Audience Member Is Separated by at Least A GenerationAnonymous RAvenNessuna valutazione finora
- Puc18 Bioengineering Lab ReportDocumento9 paginePuc18 Bioengineering Lab ReportAnonymous RAvenNessuna valutazione finora
- Valvular Heart DiseaseDocumento2 pagineValvular Heart DiseaseAnonymous RAvenNessuna valutazione finora
- Physics Exam Practice IIDocumento10 paginePhysics Exam Practice IIAnonymous RAvenNessuna valutazione finora
- Bio Lab - Respiration and Mitosis NotesDocumento2 pagineBio Lab - Respiration and Mitosis NotesAnonymous RAvenNessuna valutazione finora
- Essay On School AttendanceDocumento7 pagineEssay On School AttendanceAnonymous RAvenNessuna valutazione finora
- Datau 32Documento3 pagineDatau 32Anonymous RAvenNessuna valutazione finora
- Datau 32Documento3 pagineDatau 32Anonymous RAvenNessuna valutazione finora
- PrelabDocumento4 paginePrelabAnonymous RAvenNessuna valutazione finora
- Computer Chemistry Assignment: 2 4 (Aq) + (Aq) 4 - (Aq)Documento2 pagineComputer Chemistry Assignment: 2 4 (Aq) + (Aq) 4 - (Aq)Anonymous RAvenNessuna valutazione finora
- My Twisted World - Elliot RodgerDocumento141 pagineMy Twisted World - Elliot RodgerSusan Duclos100% (7)
- Lecture 9 Fall 2011v1Documento29 pagineLecture 9 Fall 2011v1Anonymous RAvenNessuna valutazione finora
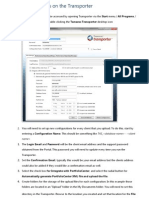
- TransporterDocumento11 pagineTransporterAnonymous RAvenNessuna valutazione finora
- pET System Manual, 11th EditionDocumento80 paginepET System Manual, 11th Editionliang2012Nessuna valutazione finora
- Plant Biotechnology For Crop ImprovementDocumento22 paginePlant Biotechnology For Crop ImprovementMint PepperNessuna valutazione finora
- Aniket Sengupta CV 2022 JanDocumento8 pagineAniket Sengupta CV 2022 Janapi-343430641Nessuna valutazione finora
- ChymosinDocumento242 pagineChymosinvbortizlNessuna valutazione finora
- The Process of Genetic EngineeringDocumento21 pagineThe Process of Genetic EngineeringJam Gumanoy NavarroNessuna valutazione finora
- Basic Concepts of Gene CloningDocumento100 pagineBasic Concepts of Gene Cloningapi-3839553100% (3)
- Transformation and ImmortalizationDocumento24 pagineTransformation and ImmortalizationSheerin SulthanaNessuna valutazione finora
- Bacterial Genetic SystemDocumento13 pagineBacterial Genetic SystemKaiyama AkhtarNessuna valutazione finora
- Perpetuation of LifeDocumento15 paginePerpetuation of LifeMomo Capoquian100% (1)
- BT Brinjal Case StudyDocumento26 pagineBT Brinjal Case StudyVeerendra Singh NagoriaNessuna valutazione finora
- MCQ MicrobiologyDocumento126 pagineMCQ MicrobiologyNohaOmar100% (2)
- Lecture 6-Gene CloningDocumento43 pagineLecture 6-Gene CloningNurfarahain ZolkeflyNessuna valutazione finora
- Cloning and Expression of Foreign Genes in E.ColiDocumento12 pagineCloning and Expression of Foreign Genes in E.ColiADITYAROOP PATHAKNessuna valutazione finora
- Bioprocess 1 SBT 2132Documento13 pagineBioprocess 1 SBT 2132Biotechnology IIUM Kuantan100% (2)
- Gene Transfer Methods MHRD NotesDocumento38 pagineGene Transfer Methods MHRD Notessrinivas ceoNessuna valutazione finora
- Quiz MicrobiologyDocumento65 pagineQuiz MicrobiologyMedShare98% (51)
- Leva DuraDocumento31 pagineLeva DuraYamile RodriguezNessuna valutazione finora
- Cloning Vector: Effat Jahan TamannaDocumento35 pagineCloning Vector: Effat Jahan TamannaHarsh vardhanNessuna valutazione finora
- Study On The Transformation Codes StuffDocumento2 pagineStudy On The Transformation Codes StuffManojkumar NairNessuna valutazione finora
- Aplikasi Transfer Gen Acc Oksidase Dalam Proses Penundaan Pemasakan Buah Pepaya (Carica Papaya L.)Documento8 pagineAplikasi Transfer Gen Acc Oksidase Dalam Proses Penundaan Pemasakan Buah Pepaya (Carica Papaya L.)Wakiatil RosidaNessuna valutazione finora
- Ligation, Expression, and Cloning of EGFP ProteinDocumento16 pagineLigation, Expression, and Cloning of EGFP ProteincupchungaiNessuna valutazione finora
- Bacterial Transformation LabDocumento23 pagineBacterial Transformation Labapi-520142437Nessuna valutazione finora
- Lecture 06 - Tools and Techniques in BiotechnologyDocumento162 pagineLecture 06 - Tools and Techniques in BiotechnologyAlkhair SangcopanNessuna valutazione finora
- History of Coffee Board at ThandikudiDocumento45 pagineHistory of Coffee Board at ThandikudiGuruprasath J100% (1)
- Gene: Fine Structure of GeneDocumento92 pagineGene: Fine Structure of GeneAshrita PradhanNessuna valutazione finora
- Gene Transfer TechniquesDocumento36 pagineGene Transfer TechniquesDrMumtaz F MusaliarNessuna valutazione finora
- ) The Right Answer: Malwanchal UniversityDocumento3 pagine) The Right Answer: Malwanchal Universityswapnil goyalNessuna valutazione finora
- Virus InfluenzaDocumento6 pagineVirus InfluenzatiaNessuna valutazione finora
- Micro para - CompiledDocumento94 pagineMicro para - CompiledJanie-Vi GorospeNessuna valutazione finora
- Module 1 - Central DogmaDocumento8 pagineModule 1 - Central DogmaAnanya SinghNessuna valutazione finora
- Taste: Surprising Stories and Science About Why Food Tastes GoodDa EverandTaste: Surprising Stories and Science About Why Food Tastes GoodValutazione: 3 su 5 stelle3/5 (20)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeDa EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeValutazione: 5 su 5 stelle5/5 (1)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactDa EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactValutazione: 5 su 5 stelle5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincDa EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincValutazione: 3.5 su 5 stelle3.5/5 (137)
- It's Elemental: The Hidden Chemistry in EverythingDa EverandIt's Elemental: The Hidden Chemistry in EverythingValutazione: 4 su 5 stelle4/5 (10)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeDa EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeValutazione: 5 su 5 stelle5/5 (4)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeDa EverandChemistry for Breakfast: The Amazing Science of Everyday LifeValutazione: 4.5 su 5 stelle4.5/5 (14)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeDa EverandChemistry for Breakfast: The Amazing Science of Everyday LifeValutazione: 4.5 su 5 stelle4.5/5 (90)
- Tribology: Friction and Wear of Engineering MaterialsDa EverandTribology: Friction and Wear of Engineering MaterialsValutazione: 5 su 5 stelle5/5 (1)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeDa EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNessuna valutazione finora
- Guidelines for Defining Process Safety Competency RequirementsDa EverandGuidelines for Defining Process Safety Competency RequirementsValutazione: 3 su 5 stelle3/5 (1)
- The Periodic Table: A Very Short IntroductionDa EverandThe Periodic Table: A Very Short IntroductionValutazione: 4.5 su 5 stelle4.5/5 (3)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeDa EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeValutazione: 4 su 5 stelle4/5 (1)
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideDa EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNessuna valutazione finora
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsDa EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsValutazione: 4 su 5 stelle4/5 (146)
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsDa EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsNessuna valutazione finora
- Ingredients: A Visual Exploration of 75 Additives & 25 Food ProductsDa EverandIngredients: A Visual Exploration of 75 Additives & 25 Food ProductsValutazione: 4 su 5 stelle4/5 (1)
- Chemistry: a QuickStudy Laminated Reference GuideDa EverandChemistry: a QuickStudy Laminated Reference GuideValutazione: 5 su 5 stelle5/5 (1)
- Phase Equilibria in Chemical EngineeringDa EverandPhase Equilibria in Chemical EngineeringValutazione: 4 su 5 stelle4/5 (11)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolDa EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNessuna valutazione finora