Potrebbero piacerti anche
- Cleaning Validation SOP Novartis PDFDocumento15 pagineCleaning Validation SOP Novartis PDFFaisal Abbas100% (2)
- Good Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsDa EverandGood Manufacturing Practices (GMP) Modules for Pharmaceutical ProductsNessuna valutazione finora
- GMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsDa EverandGMP in Pharmaceutical Industry: Global cGMP & Regulatory ExpectationsValutazione: 5 su 5 stelle5/5 (2)
- Cleaning Validation Lifecycle ApproachDocumento50 pagineCleaning Validation Lifecycle ApproachlmqasemNessuna valutazione finora
- Validation of Coating Equipment (Ketik Ulang)Documento6 pagineValidation of Coating Equipment (Ketik Ulang)Dedhieaja0% (1)
- Media Fill ChecklistDocumento11 pagineMedia Fill ChecklistSilke Igemann100% (1)
- CCS Guide Attachment3 v2Documento34 pagineCCS Guide Attachment3 v2elisabetta ghilardi100% (1)
- Mobile Fire Extinguishers. Characteristics, Performance and Test MethodsDocumento28 pagineMobile Fire Extinguishers. Characteristics, Performance and Test MethodsSawita LertsupochavanichNessuna valutazione finora
- Paul S. Adler - Paul Du Gay - Glenn Morgan - Michael Reed (Eds.) - The Oxford Handbook of Sociology, Social Theory, and Organization Studies - Contemporary Currents-Oxford University Press, USA (2014)Documento817 paginePaul S. Adler - Paul Du Gay - Glenn Morgan - Michael Reed (Eds.) - The Oxford Handbook of Sociology, Social Theory, and Organization Studies - Contemporary Currents-Oxford University Press, USA (2014)Andreea Dobrita67% (3)
- Cleaning Validation in Pharmaceutical IndustryDocumento21 pagineCleaning Validation in Pharmaceutical IndustrysvengotoNessuna valutazione finora
- Cleaning Validation Guideline SampleDocumento3 pagineCleaning Validation Guideline SampleSagi NguyenNessuna valutazione finora
- ValidationDocumento56 pagineValidationAmit Singh100% (1)
- Pharmaceutical Cleaning A Comprehensive Approach - 0Documento15 paginePharmaceutical Cleaning A Comprehensive Approach - 0Mina Maher MikhailNessuna valutazione finora
- Validation in Pharmaceutical Manufacturing IndustryDocumento5 pagineValidation in Pharmaceutical Manufacturing IndustryNAVNEET BAGGANessuna valutazione finora
- Biocontamination Control for Pharmaceuticals and HealthcareDa EverandBiocontamination Control for Pharmaceuticals and HealthcareValutazione: 5 su 5 stelle5/5 (1)
- Cleanroom Technology: Fundamentals of Design, Testing and OperationDa EverandCleanroom Technology: Fundamentals of Design, Testing and OperationNessuna valutazione finora
- Pharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersDa EverandPharmaceutical Industry Documents: 90 Pharmaceutical Quality Assurance Interview Questions & AnswersNessuna valutazione finora
- Risk Management Applications in Pharmaceutical and Biopharmaceutical ManufacturingDa EverandRisk Management Applications in Pharmaceutical and Biopharmaceutical ManufacturingHamid MollahNessuna valutazione finora
- Practical Approaches to Method Validation and Essential Instrument QualificationDa EverandPractical Approaches to Method Validation and Essential Instrument QualificationNessuna valutazione finora
- Pharmaceutical Quality Management System (QMS) Questions and AnswersDa EverandPharmaceutical Quality Management System (QMS) Questions and AnswersNessuna valutazione finora
- Cleaning Validation MACO v2.0Documento2 pagineCleaning Validation MACO v2.0Ovais08100% (2)
- Cleaning Validation and Its Importance in Pharmaceutical IndustryDocumento5 pagineCleaning Validation and Its Importance in Pharmaceutical Industrymichael_payne3532Nessuna valutazione finora
- Overview of Cleaning Validation in Pharmaceutical Manufacturing Unit PDFDocumento11 pagineOverview of Cleaning Validation in Pharmaceutical Manufacturing Unit PDFAlexander YVNessuna valutazione finora
- Hold Time Study Protocol OF Cleaned Manufacturing Equipment Awaiting For UseDocumento11 pagineHold Time Study Protocol OF Cleaned Manufacturing Equipment Awaiting For Usegopusankar100% (5)
- Risk Assesment For Cleaning ValidationDocumento11 pagineRisk Assesment For Cleaning Validationqfbfabyhola100% (4)
- Risk Assessment and Management For Healthcare Manufacturing: Practical Tips and Case StudiesDocumento14 pagineRisk Assessment and Management For Healthcare Manufacturing: Practical Tips and Case StudiesTim SandleNessuna valutazione finora
- Master Cleaning Validation PlanDocumento25 pagineMaster Cleaning Validation PlanWidya Lukitasari100% (1)
- Cleaning Validation Rinsing TesDocumento5 pagineCleaning Validation Rinsing TesUrsula HilleNessuna valutazione finora
- Procedurefor Cleaning ValidationDocumento21 pagineProcedurefor Cleaning ValidationQ CNessuna valutazione finora
- Cleaning ValidationDocumento41 pagineCleaning Validationvipin_chaudhary93% (14)
- Life Cycle Risk Assessment of HPLC InstrumentsDocumento13 pagineLife Cycle Risk Assessment of HPLC Instrumentsalexg123100% (1)
- Cleaning Validation Sample ProtocolDocumento7 pagineCleaning Validation Sample ProtocolArieTamaNessuna valutazione finora
- Establishing A Complete Set of Target, Alert, and Action Limits For Microbial Counts in Purified WaterDocumento8 pagineEstablishing A Complete Set of Target, Alert, and Action Limits For Microbial Counts in Purified WaterSagi Nguyen100% (2)
- Setting Limits For Use in Cleaning ValidationDocumento65 pagineSetting Limits For Use in Cleaning ValidationmaraperezlindoNessuna valutazione finora
- Batch DispositionDocumento21 pagineBatch DispositionNUPharma Consular100% (1)
- Cleaning Validation ProtocolDocumento9 pagineCleaning Validation Protocolyash143565100% (2)
- Cleaning Validation StudyDocumento12 pagineCleaning Validation StudyG_Ranjith100% (3)
- Aseptic Process ValidationDocumento32 pagineAseptic Process ValidationG_Ranjith100% (1)
- Process Validation FDADocumento40 pagineProcess Validation FDALarissa Goncalves100% (1)
- Process Validation of LiquidDocumento24 pagineProcess Validation of LiquidAshutosh Shukla100% (2)
- Calculation of Acceptable Residue Limits GeneralDocumento2 pagineCalculation of Acceptable Residue Limits Generaljljimenez1969100% (1)
- Texwipe PDA Cleaning and Cleaning Validation Chapter19Documento26 pagineTexwipe PDA Cleaning and Cleaning Validation Chapter19davincicode888100% (1)
- BioburdenDocumento95 pagineBioburdenTumma RamaraoNessuna valutazione finora
- Validation-An Important Tool of GMP: About Authors: Karmveer TomarDocumento4 pagineValidation-An Important Tool of GMP: About Authors: Karmveer TomarShiv KumarNessuna valutazione finora
- Water Alert Action Limits PramodthDocumento12 pagineWater Alert Action Limits Pramodthvenkat_du2000100% (2)
- Annex4-TRS992 Hold Time Study GuidelineDocumento8 pagineAnnex4-TRS992 Hold Time Study Guidelinensk79in@gmail.com100% (1)
- Iqoqpq RMGDocumento11 pagineIqoqpq RMGjpmaurya7750% (4)
- Media Fill FDA 483 ObservationsDocumento22 pagineMedia Fill FDA 483 Observationsvijayns_250355172Nessuna valutazione finora
- Particle Monitoring Requirements in Pharmaceutical CleanroomsDocumento7 pagineParticle Monitoring Requirements in Pharmaceutical CleanroomsananthNessuna valutazione finora
- Sampling Water Table ContentsDocumento2 pagineSampling Water Table Contentsraju1559405Nessuna valutazione finora
- Clenaing ValidationDocumento31 pagineClenaing Validationagarciah15891100% (2)
- 04 ISPEs Guides and How They Apply To Cleaning and Cleaning Validation by Stephanie WilkinsDocumento33 pagine04 ISPEs Guides and How They Apply To Cleaning and Cleaning Validation by Stephanie Wilkinsmjamil0995100% (1)
- 2-4 ProcessValidationDocumento37 pagine2-4 ProcessValidationlouish9175841100% (2)
- Microbial Risk Assesment in Pharmaceutical CleanroomsDocumento9 pagineMicrobial Risk Assesment in Pharmaceutical CleanroomsteaNessuna valutazione finora
- Validation Master Plan A Complete Guide - 2020 EditionDa EverandValidation Master Plan A Complete Guide - 2020 EditionNessuna valutazione finora
- Regulatory Aspects of Pharmaceutical Quality System: Brief IntroductionDa EverandRegulatory Aspects of Pharmaceutical Quality System: Brief IntroductionNessuna valutazione finora
- Narrow Vs BroadDocumento12 pagineNarrow Vs Broadssrz7786Nessuna valutazione finora
- $Ssuryhg5Hylvhg - DQ 0dwhuldo 6'61XPEHU 9huvlrq: 3hufhqwdjh &$651, QjuhglhqwvDocumento6 pagine$Ssuryhg5Hylvhg - DQ 0dwhuldo 6'61XPEHU 9huvlrq: 3hufhqwdjh &$651, Qjuhglhqwvssrz7786Nessuna valutazione finora
- 0803mar 40Documento1 pagina0803mar 40ssrz7786Nessuna valutazione finora
- GalenIQ™ 981Documento2 pagineGalenIQ™ 981ssrz7786Nessuna valutazione finora
- Zakat Calculation SpreadsheetDocumento6 pagineZakat Calculation SpreadsheetelmechanaNessuna valutazione finora
- Income Tax Return E-Filing Guide (Salaried) - 2009Documento15 pagineIncome Tax Return E-Filing Guide (Salaried) - 2009ssrz7786Nessuna valutazione finora
- A Comprehensive Review On Renewable and Sustainable Heating Systems For Poultry FarmingDocumento22 pagineA Comprehensive Review On Renewable and Sustainable Heating Systems For Poultry FarmingPl TorrNessuna valutazione finora
- User Custom PP Install74Documento2 pagineUser Custom PP Install74Zixi FongNessuna valutazione finora
- FC2060Documento10 pagineFC2060esnNessuna valutazione finora
- WS-250 4BB 60 Cells 40mm DatasheetDocumento2 pagineWS-250 4BB 60 Cells 40mm DatasheetTejash NaikNessuna valutazione finora
- Mio Digiwalker c220/c220sDocumento32 pagineMio Digiwalker c220/c220sTNessuna valutazione finora
- Joseph J. Fiumara v. Fireman's Fund Insurance Companies, 746 F.2d 87, 1st Cir. (1984)Documento7 pagineJoseph J. Fiumara v. Fireman's Fund Insurance Companies, 746 F.2d 87, 1st Cir. (1984)Scribd Government DocsNessuna valutazione finora
- General Mathematics 2nd Quarter ExamDocumento3 pagineGeneral Mathematics 2nd Quarter ExamDeped TambayanNessuna valutazione finora
- CIVREV!!!!Documento5 pagineCIVREV!!!!aypod100% (1)
- Suggested Answers Spring 2015 Examinations 1 of 8: Strategic Management Accounting - Semester-6Documento8 pagineSuggested Answers Spring 2015 Examinations 1 of 8: Strategic Management Accounting - Semester-6Abdul BasitNessuna valutazione finora
- What Is EBSD ? Why Use EBSD ? Why Measure Microstructure ? What Does EBSD Do That Cannot Already Be Done ?Documento5 pagineWhat Is EBSD ? Why Use EBSD ? Why Measure Microstructure ? What Does EBSD Do That Cannot Already Be Done ?Zahir Rayhan JhonNessuna valutazione finora
- Release ACOS 4.1.4-GR1-P10 IssuesDocumento241 pagineRelease ACOS 4.1.4-GR1-P10 IssuesdanielatellaNessuna valutazione finora
- Induction-Llgd 2022Documento11 pagineInduction-Llgd 2022Phạm Trúc QuỳnhNessuna valutazione finora
- Grade 5 Olympiad: Answer The QuestionsDocumento14 pagineGrade 5 Olympiad: Answer The QuestionsVinieysha LoganathanNessuna valutazione finora
- NGOs in Satkhira PresentationDocumento17 pagineNGOs in Satkhira PresentationRubayet KhundokerNessuna valutazione finora
- West Bengal Joint Entrance Examinations Board: Provisional Admission LetterDocumento2 pagineWest Bengal Joint Entrance Examinations Board: Provisional Admission Lettertapas chakrabortyNessuna valutazione finora
- TC 9-237 Welding 1993Documento680 pagineTC 9-237 Welding 1993enricoNessuna valutazione finora
- SMPLEDocumento2 pagineSMPLEKla AlvarezNessuna valutazione finora
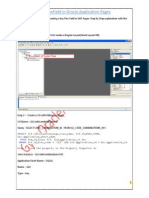
- KFF in OAF Page-GyanDocumento4 pagineKFF in OAF Page-Gyangyan darpanNessuna valutazione finora
- Stress and Strain - Axial LoadingDocumento18 pagineStress and Strain - Axial LoadingClackfuik12Nessuna valutazione finora
- Aug 2020 Builders Line Tamil MonthlyDocumento48 pagineAug 2020 Builders Line Tamil MonthlyBuildersLineMonthlyNessuna valutazione finora
- Current Matching Control System For Multi-Terminal DC Transmission To Integrate Offshore Wind FarmsDocumento6 pagineCurrent Matching Control System For Multi-Terminal DC Transmission To Integrate Offshore Wind FarmsJackie ChuNessuna valutazione finora
- Reading Task CardsDocumento2 pagineReading Task CardscatnappleNessuna valutazione finora
- LG LCD TV 32lp1dc - Al-04ca Service ManualDocumento47 pagineLG LCD TV 32lp1dc - Al-04ca Service ManualJavin GallardoNessuna valutazione finora
- Project Cost ContingencyDocumento9 pagineProject Cost ContingencyniroshniroshNessuna valutazione finora
- Panch ShilDocumento118 paginePanch ShilSohel BangiNessuna valutazione finora
- UNECE-Turkey-TCDO-Rail Freight Traffic in Euro-Asian LinksDocumento20 pagineUNECE-Turkey-TCDO-Rail Freight Traffic in Euro-Asian LinksArseneNessuna valutazione finora
- T53 L 13 Turboshaft EngineDocumento2 pagineT53 L 13 Turboshaft EngineEagle1968Nessuna valutazione finora
- Hi 3 Yt 318201Documento3 pagineHi 3 Yt 318201partha khatuaNessuna valutazione finora