Potrebbero piacerti anche
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5795)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- UBKV Ranking Proforma With Annexures 2018 PDFDocumento53 pagineUBKV Ranking Proforma With Annexures 2018 PDFSubinay Saha RoyNessuna valutazione finora
- Ethnomedicinal Plants For Indigestion in Uthiramerur Taluk Kancheepuram District Tamilnadu IndiaDocumento8 pagineEthnomedicinal Plants For Indigestion in Uthiramerur Taluk Kancheepuram District Tamilnadu IndiaGladys DjeugaNessuna valutazione finora
- Internationalresidential Code 2009 Edition Fuel Gas SectionDocumento49 pagineInternationalresidential Code 2009 Edition Fuel Gas SectionZarex BorjaNessuna valutazione finora
- Ce Mark - Application FormDocumento3 pagineCe Mark - Application Formrajivsinghal90Nessuna valutazione finora
- Design and Details of Elevated Steel Tank PDFDocumento10 pagineDesign and Details of Elevated Steel Tank PDFandysupaNessuna valutazione finora
- Jebao DCP Pump User ManualDocumento3 pagineJebao DCP Pump User ManualSubrata Das100% (1)
- Mon AnhDocumento7 pagineMon AnhDavid NguyenNessuna valutazione finora
- Practice Test For Exam 3 Name: Miguel Vivas Score: - /10Documento2 paginePractice Test For Exam 3 Name: Miguel Vivas Score: - /10MIGUEL ANGELNessuna valutazione finora
- Course Weekly Schedule Health Science TheoryDocumento6 pagineCourse Weekly Schedule Health Science Theoryapi-466810096Nessuna valutazione finora
- Musa Paradisiaca L. and Musa Sapientum L.: A Phytochemical and Pharmacological ReviewDocumento8 pagineMusa Paradisiaca L. and Musa Sapientum L.: A Phytochemical and Pharmacological ReviewDeviNessuna valutazione finora
- PYMS Is A Reliable Malnutrition Screening ToolsDocumento8 paginePYMS Is A Reliable Malnutrition Screening ToolsRika LedyNessuna valutazione finora
- Plastic Omnium 2015 RegistrationDocumento208 paginePlastic Omnium 2015 Registrationgsravan_23Nessuna valutazione finora
- Five Characteristics of Authentic LeadershipDocumento2 pagineFive Characteristics of Authentic LeadershipArnel Billy LimNessuna valutazione finora
- EESC 111 Worksheets Module 5Documento5 pagineEESC 111 Worksheets Module 5Keira O'HowNessuna valutazione finora
- Leave of Absence Form (Rev. 02 072017)Documento1 paginaLeave of Absence Form (Rev. 02 072017)KIMBERLY BALISACANNessuna valutazione finora
- MINUZA Laptop Scheme Programs ThyDocumento9 pagineMINUZA Laptop Scheme Programs Thyanualithe kamalizaNessuna valutazione finora
- Industrial Visit ReportDocumento8 pagineIndustrial Visit ReportAnuragBoraNessuna valutazione finora
- 1 SMDocumento10 pagine1 SMAnindita GaluhNessuna valutazione finora
- Contemporary ImageDocumento43 pagineContemporary ImageProf. L100% (1)
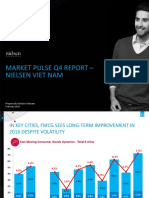
- Market Pulse Q4 Report - Nielsen Viet Nam: Prepared by Nielsen Vietnam February 2017Documento8 pagineMarket Pulse Q4 Report - Nielsen Viet Nam: Prepared by Nielsen Vietnam February 2017K57.CTTT BUI NGUYEN HUONG LYNessuna valutazione finora
- Learning Activity Sheet General Chemistry 2 (Q4 - Lessons 7 and 8) Electrochemical ReactionsDocumento11 pagineLearning Activity Sheet General Chemistry 2 (Q4 - Lessons 7 and 8) Electrochemical Reactionsprincess3canlasNessuna valutazione finora
- The Human Body: An Orientation: Part ADocumento10 pagineThe Human Body: An Orientation: Part ARoi Christoffer Jocson PeraltaNessuna valutazione finora
- Pineapple PDFDocumento7 paginePineapple PDFDestia AyuNessuna valutazione finora
- Silly VersesDocumento29 pagineSilly Verseskevin daleNessuna valutazione finora
- R. Nishanth K. VigneswaranDocumento20 pagineR. Nishanth K. VigneswaranAbishaTeslinNessuna valutazione finora
- IWCF Comb. Supv Equip. 01Documento25 pagineIWCF Comb. Supv Equip. 01andrzema100% (3)
- 2015 4-H Show & Sale CatalogDocumento53 pagine2015 4-H Show & Sale CatalogFauquier NowNessuna valutazione finora
- Material: Safety Data SheetDocumento3 pagineMaterial: Safety Data SheetMichael JoudalNessuna valutazione finora
- GeoSS Event Seminar 12 July 2012 - SlidesDocumento15 pagineGeoSS Event Seminar 12 July 2012 - SlidesNurmanda RamadhaniNessuna valutazione finora
- Plyometric Training: Sports Med 2Documento9 paginePlyometric Training: Sports Med 2Viren ManiyarNessuna valutazione finora