Potrebbero piacerti anche
- Physical Organic Notes 9pDocumento9 paginePhysical Organic Notes 9pprincejoes194Nessuna valutazione finora
- Use of Isotopes For Studying Reaction Mechanisms: - JU-n-e-1-9-9-7 - 4-7Documento7 pagineUse of Isotopes For Studying Reaction Mechanisms: - JU-n-e-1-9-9-7 - 4-7Ab AbNessuna valutazione finora
- Kinetic Isotope Effect in Organic ChemistryDocumento28 pagineKinetic Isotope Effect in Organic ChemistryMaximiliano DelahigueraNessuna valutazione finora
- Isotope 3Documento6 pagineIsotope 3Ashish Manatosh BarikNessuna valutazione finora
- Vibration - Rotation Spectroscopy of HCL and DCLDocumento9 pagineVibration - Rotation Spectroscopy of HCL and DCLAngela LamasNessuna valutazione finora
- Collision Theory and Transition State TheoryDocumento29 pagineCollision Theory and Transition State Theorysmi_santhoshNessuna valutazione finora
- Heat: The Nature of Temperature and Most Other PhysicsDa EverandHeat: The Nature of Temperature and Most Other PhysicsNessuna valutazione finora
- Introduction to Physical Organic Chemistry Thermodynamics and KineticsDocumento11 pagineIntroduction to Physical Organic Chemistry Thermodynamics and KineticsAcidri Abdulkarim100% (1)
- Belousov-Zhabotinskii Reaction: Physical Chemistry Laboratory ExperimentDocumento27 pagineBelousov-Zhabotinskii Reaction: Physical Chemistry Laboratory ExperimentLetitia SarahNessuna valutazione finora
- Inverse Kinetic Isotope Effect ExplainedDocumento5 pagineInverse Kinetic Isotope Effect ExplainedAlessandro Da Cruz GoncalvesNessuna valutazione finora
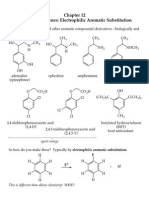
- Chapter 12Documento23 pagineChapter 12Hải NguyễnNessuna valutazione finora
- 11.2 Intermolecular ForcesDocumento10 pagine11.2 Intermolecular ForcesethanNessuna valutazione finora
- Chem 481 C 14Documento6 pagineChem 481 C 14Bayram KarahanNessuna valutazione finora
- GOC NotesDocumento43 pagineGOC NotesSudhanshu HedaNessuna valutazione finora
- Topic 7a - Bimolecular ReactionsDocumento12 pagineTopic 7a - Bimolecular ReactionsChristine Pui YiNessuna valutazione finora
- Chapter7elimination Ans SubstnDocumento22 pagineChapter7elimination Ans Substnjagabandhu_patraNessuna valutazione finora
- Lec 10Documento3 pagineLec 10Pratik HoraNessuna valutazione finora
- Emission and recombination coefficients for hydrogen with κ-distributed electron energiesDocumento3 pagineEmission and recombination coefficients for hydrogen with κ-distributed electron energiesjameswhite4321Nessuna valutazione finora
- Chem 002 Angel C. de Dios: ElectrochemistryDocumento5 pagineChem 002 Angel C. de Dios: ElectrochemistryBenni WewokNessuna valutazione finora
- Kinetics III Exercises - With SolutionsDocumento20 pagineKinetics III Exercises - With SolutionsPratham JhaNessuna valutazione finora
- S. Stamatiadis Et Al - Saddle-Node States in The Spectra of HCO and DCO: A Periodic Orbit Classification of Vibrational LevelsDocumento8 pagineS. Stamatiadis Et Al - Saddle-Node States in The Spectra of HCO and DCO: A Periodic Orbit Classification of Vibrational LevelsMaxnamewNessuna valutazione finora
- S. Aubry - Bipolaronic Charge Density Waves and Polaronic Spin Density Waves in The Holstein-Hubbard ModelDocumento7 pagineS. Aubry - Bipolaronic Charge Density Waves and Polaronic Spin Density Waves in The Holstein-Hubbard ModelGlade680Nessuna valutazione finora
- Electrochemical Kinetics of Corrosion and Passivity (EKCPDocumento36 pagineElectrochemical Kinetics of Corrosion and Passivity (EKCPWan Ibrahim Al-KutaniNessuna valutazione finora
- Comparison of Equilibrium Constants in Gas and Liquid PhasesDocumento6 pagineComparison of Equilibrium Constants in Gas and Liquid Phaseswesileh981Nessuna valutazione finora
- Experiment 6Documento7 pagineExperiment 6sajithNessuna valutazione finora
- Chemistry XI - Part-1Documento4 pagineChemistry XI - Part-1Janki VernekarNessuna valutazione finora
- NMR N M R: Uclear Agnetic EsonanceDocumento33 pagineNMR N M R: Uclear Agnetic EsonanceCarolina AlpucheNessuna valutazione finora
- CHM2046 Chapter 11Documento64 pagineCHM2046 Chapter 11wbryant2013Nessuna valutazione finora
- Byrnes Phys. Rev. Lett. 105, 186402 (2010)Documento4 pagineByrnes Phys. Rev. Lett. 105, 186402 (2010)Tim ByrnesNessuna valutazione finora
- Answers To Assignment2-2009Documento6 pagineAnswers To Assignment2-2009ElmIeyHakiemiEyNessuna valutazione finora
- SCO VibrationSurfacesDocumento2 pagineSCO VibrationSurfacesSkintone PhotographyNessuna valutazione finora
- Overcoming Limitations of Hartree-Fock for Electron CorrelationDocumento7 pagineOvercoming Limitations of Hartree-Fock for Electron CorrelationDiego Alejandro Hurtado BalcazarNessuna valutazione finora
- Chem 232 Fall 2015 UIUC NotesDocumento274 pagineChem 232 Fall 2015 UIUC NotesSri KondabattulaNessuna valutazione finora
- GR XI Term 2 CHEMISTRY Ans KeyDocumento10 pagineGR XI Term 2 CHEMISTRY Ans Keyrohan fernandesNessuna valutazione finora
- Spectroscopy I (1a)Documento6 pagineSpectroscopy I (1a)shaharhr1Nessuna valutazione finora
- Lecture 6 Kinetic Isotope EffectDocumento11 pagineLecture 6 Kinetic Isotope EffectcsnNessuna valutazione finora
- Lecture 1Documento11 pagineLecture 1Fang GaoNessuna valutazione finora
- CH2416 - BKC Revision NotesDocumento2 pagineCH2416 - BKC Revision NotesJohnNessuna valutazione finora
- KineticsDocumento51 pagineKineticsSaumil Sachdeva100% (1)
- Approximate Lcao Molecular Orbital TheoryDocumento9 pagineApproximate Lcao Molecular Orbital TheoryJack RyderNessuna valutazione finora
- Httpspmt.physicsandmathstutor.comdownloadChemistryA LevelNotesOCR A3 Periodic Table and EnergyDetailed3.2.20PhysicalDocumento17 pagineHttpspmt.physicsandmathstutor.comdownloadChemistryA LevelNotesOCR A3 Periodic Table and EnergyDetailed3.2.20Physicalz akterNessuna valutazione finora
- Electrochem NotesDocumento14 pagineElectrochem NotesKaitlynNessuna valutazione finora
- Random Physics ArticlesDocumento44 pagineRandom Physics ArticlesAzam-Savaşçı Anderson MohammadNessuna valutazione finora
- Teori VB LanjutDocumento24 pagineTeori VB LanjutZonasalsa ArdyantiNessuna valutazione finora
- Chemical EnergeticsDocumento6 pagineChemical EnergeticsahumanbeinginearthNessuna valutazione finora
- NMR Lecture 4 Chemical ShiftDocumento15 pagineNMR Lecture 4 Chemical ShiftAnselmo Mtz GagosNessuna valutazione finora
- Chapter 16 - Section B - Non-Numerical SolutionsDocumento3 pagineChapter 16 - Section B - Non-Numerical SolutionsKhalid M MohammedNessuna valutazione finora
- HCI 2021 Prelim Paper 1 SolutionsDocumento18 pagineHCI 2021 Prelim Paper 1 Solutions4A730RudhreshNessuna valutazione finora
- Second Quantization and Elementary Excitations in SolidsDocumento5 pagineSecond Quantization and Elementary Excitations in SolidsbonzongNessuna valutazione finora
- Frick Chemical Laboratory contribution on dipole momentsDocumento14 pagineFrick Chemical Laboratory contribution on dipole momentsAnonymous FigYuONxuuNessuna valutazione finora
- 226 Alkene Rxns LecDocumento12 pagine226 Alkene Rxns LecKaaya GodfreyNessuna valutazione finora
- Statistical Thermodynamics of Lattice Gases and Electrochemical PotentialDocumento7 pagineStatistical Thermodynamics of Lattice Gases and Electrochemical PotentialNaveen NaviNessuna valutazione finora
- CH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHDocumento8 pagineCH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHShahbaz NazirNessuna valutazione finora
- Electronic Correlation: Problems With HF-SCF Polyelectronic FunctionsDocumento26 pagineElectronic Correlation: Problems With HF-SCF Polyelectronic FunctionsAnonymous rn2qoBPjKyNessuna valutazione finora
- Ion Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsDa EverandIon Association in Proton Transfer Reactions: Use of ESR for the Quantitative Determination of Gas Phase Atom and Radical ConcentrationsNessuna valutazione finora
- Feynman Lectures Simplified 3B: Quantum Mechanics Part TwoDa EverandFeynman Lectures Simplified 3B: Quantum Mechanics Part TwoNessuna valutazione finora
- Reactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsDa EverandReactive Oxygen Species: Signaling Between Hierarchical Levels in PlantsFranz-Josef SchmittNessuna valutazione finora
- Fundamental Analysis of Aviation SectorDocumento34 pagineFundamental Analysis of Aviation SectorAbhay KapkotiNessuna valutazione finora
- 3 Why Self Help FailsDocumento5 pagine3 Why Self Help FailsAbhay KapkotiNessuna valutazione finora
- 3 Why Self Help FailsDocumento5 pagine3 Why Self Help FailsAbhay KapkotiNessuna valutazione finora
- 2 Active Notes and BreaksDocumento2 pagine2 Active Notes and BreaksAbhay KapkotiNessuna valutazione finora
- Nano MaterialsDocumento13 pagineNano MaterialsAbhay KapkotiNessuna valutazione finora
- The Dehli OrdealDocumento1 paginaThe Dehli OrdealAbhay KapkotiNessuna valutazione finora
- Fundamental Analysis of PETROLEUM SectorDocumento31 pagineFundamental Analysis of PETROLEUM SectorAbhay Kapkoti0% (1)
- Schools Are PrisonDocumento2 pagineSchools Are PrisonAbhay KapkotiNessuna valutazione finora
- CANVAS 2015 SPONSORSHIP PACKAGEDocumento2 pagineCANVAS 2015 SPONSORSHIP PACKAGEAbhay KapkotiNessuna valutazione finora
- International Trade and Capital FlowsDocumento48 pagineInternational Trade and Capital FlowsAbhay KapkotiNessuna valutazione finora
- Supply Chain ManagementDocumento27 pagineSupply Chain ManagementskibcobsaivigneshNessuna valutazione finora
- Kumaon 6 NDocumento2 pagineKumaon 6 NAbhay KapkotiNessuna valutazione finora
- Jyoti Final TRM PaperDocumento15 pagineJyoti Final TRM PaperAbhay KapkotiNessuna valutazione finora
- Muk Tesh WarDocumento1 paginaMuk Tesh WarAbhay KapkotiNessuna valutazione finora
- Indusrty Finance IndiaDocumento56 pagineIndusrty Finance IndiaAbhay KapkotiNessuna valutazione finora
- Alumni, Placement, Call, EventDocumento1 paginaAlumni, Placement, Call, EventAbhay KapkotiNessuna valutazione finora
- On International Financial InstitutionsDocumento16 pagineOn International Financial InstitutionsYogesh Vadgaonkar0% (1)
- Neoclassical Organisation Theory Lecture on Classical vs Neoclassical ApproachesDocumento9 pagineNeoclassical Organisation Theory Lecture on Classical vs Neoclassical ApproachesAbhay KapkotiNessuna valutazione finora
- Kumayun Mandal LTDocumento406 pagineKumayun Mandal LTAbhay KapkotiNessuna valutazione finora
- Environmental Internship ExampleDocumento1 paginaEnvironmental Internship ExampleawaishaneefNessuna valutazione finora
- Munsyari: The Little KashmirDocumento4 pagineMunsyari: The Little KashmirAbhay KapkotiNessuna valutazione finora
- Caiib GBM Moda PartiiDocumento25 pagineCaiib GBM Moda Partiijayeshagiwal10Nessuna valutazione finora
- Classical & Neo Classical TheoriesDocumento29 pagineClassical & Neo Classical TheoriesAbhay Kapkoti100% (2)
- What Is InnovationDocumento7 pagineWhat Is InnovationAbhay KapkotiNessuna valutazione finora
- The Dehli OrdealDocumento1 paginaThe Dehli OrdealAbhay KapkotiNessuna valutazione finora
- Krugman Obstfeld Ch21Documento31 pagineKrugman Obstfeld Ch21Abhay KapkotiNessuna valutazione finora
- Imp FineDocumento16 pagineImp FineAbhay KapkotiNessuna valutazione finora
- Pptinsurance 130314050707 Phpapp02Documento16 paginePptinsurance 130314050707 Phpapp02Abhay KapkotiNessuna valutazione finora
- StuffDocumento1 paginaStuffAbhay KapkotiNessuna valutazione finora
- Vadiyanatha AstakamDocumento4 pagineVadiyanatha AstakamRaga MalikaNessuna valutazione finora
- Autism AspergerDocumento24 pagineAutism AspergerusavelNessuna valutazione finora
- German composer known for political worksDocumento4 pagineGerman composer known for political worksGeorge PikNessuna valutazione finora
- Web Search - One People's Public Trust 1776 UCCDocumento28 pagineWeb Search - One People's Public Trust 1776 UCCVincent J. CataldiNessuna valutazione finora
- Supplier Development at Honda, Nissan and ToyotaDocumento28 pagineSupplier Development at Honda, Nissan and Toyotapresidonsi100% (1)
- New Text DocumentDocumento8 pagineNew Text DocumentDhaniNessuna valutazione finora
- Prayer BuddyDocumento42 paginePrayer BuddyJoribelle AranteNessuna valutazione finora
- Richard CuretonDocumento24 pagineRichard CuretonHayk HambardzumyanNessuna valutazione finora
- The Role of Christian Education in Socia Group 6Documento6 pagineThe Role of Christian Education in Socia Group 6Ṭhanuama BiateNessuna valutazione finora
- MF 04Documento21 pagineMF 04Carlos De la CruzNessuna valutazione finora
- The Other Side of Love AutosavedDocumento17 pagineThe Other Side of Love AutosavedPatrick EdrosoloNessuna valutazione finora
- (Cambridge Series in Statistical and Probabilistic Mathematics) Gerhard Tutz, Ludwig-Maximilians-Universität Munchen - Regression For Categorical Data-Cambridge University Press (2012)Documento574 pagine(Cambridge Series in Statistical and Probabilistic Mathematics) Gerhard Tutz, Ludwig-Maximilians-Universität Munchen - Regression For Categorical Data-Cambridge University Press (2012)shu100% (2)
- 1022-Article Text-2961-1-10-20200120Documento10 pagine1022-Article Text-2961-1-10-20200120Zuber RokhmanNessuna valutazione finora
- Bombardier CityfloDocumento14 pagineBombardier CityfloBiju KmNessuna valutazione finora
- FunambolDocumento48 pagineFunambolAmeliaNessuna valutazione finora
- Conic SectionDocumento58 pagineConic SectionNailah Sakinah100% (1)
- Logic Puzzles Freebie: Includes Instructions!Documento12 pagineLogic Puzzles Freebie: Includes Instructions!api-507836868Nessuna valutazione finora
- Life Convict Laxman Naskar Vs State of West Bengal & Anr On 1 September, 2000Documento6 pagineLife Convict Laxman Naskar Vs State of West Bengal & Anr On 1 September, 2000Kimberly HardyNessuna valutazione finora
- Extinction - WikipediaDocumento14 pagineExtinction - Wikipediaskline3Nessuna valutazione finora
- Kurukshetra English August '17Documento60 pagineKurukshetra English August '17amit2688Nessuna valutazione finora
- Futurology and EducationDocumento32 pagineFuturology and EducationMuhammad Abubakar100% (1)
- Vinzenz Hediger, Patrick Vonderau - Films That Work - Industrial Film and The Productivity of Media (Film Culture in Transition) (2009)Documento496 pagineVinzenz Hediger, Patrick Vonderau - Films That Work - Industrial Film and The Productivity of Media (Film Culture in Transition) (2009)Arlindo Rebechi JuniorNessuna valutazione finora
- Theo 5Documento2 pagineTheo 5chingchongNessuna valutazione finora
- Homework WatergateDocumento8 pagineHomework Watergateaapsujtif100% (1)
- Guy GacottDocumento4 pagineGuy GacottAly ConcepcionNessuna valutazione finora
- Literature ReviewDocumento4 pagineLiterature Reviewapi-549241187Nessuna valutazione finora
- The Human Element is Critical in Personal SellingDocumento18 pagineThe Human Element is Critical in Personal SellingArsalan AhmedNessuna valutazione finora
- PIC16 F 1619Documento594 paginePIC16 F 1619Francisco Martinez AlemanNessuna valutazione finora
- Readingdev 7Documento2 pagineReadingdev 7api-190328610Nessuna valutazione finora