Potrebbero piacerti anche
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (344)
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (399)
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (73)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (120)
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Veterinary Gross Anatomy Female Genital TractDocumento10 pagineVeterinary Gross Anatomy Female Genital TractTatenda MagejaNessuna valutazione finora
- ALB2Documento4 pagineALB2Jonalyn SalandoNessuna valutazione finora
- Characterization of Tannia PlantDocumento104 pagineCharacterization of Tannia PlantSolomon FantawNessuna valutazione finora
- Practice Questions FinalDocumento32 paginePractice Questions FinalpsdantonioNessuna valutazione finora
- The 12 Untapped Targets To Ignite New Muscle GrowthDocumento68 pagineThe 12 Untapped Targets To Ignite New Muscle Growthace_mimNessuna valutazione finora
- Ovulation CalanderDocumento2 pagineOvulation CalanderBN NGNessuna valutazione finora
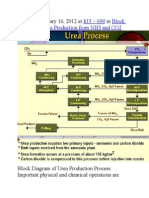
- Published January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2Documento9 paginePublished January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2himanshuchawla654Nessuna valutazione finora
- 19 - Lipid MetabolismDocumento35 pagine19 - Lipid MetabolismcheckmateNessuna valutazione finora
- Johnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160Documento20 pagineJohnson Jerry Alan Chinese Medical Qigong Therapy Vol 5-141-160toanbauNessuna valutazione finora
- ABCD Vocal Technique Handout PDFDocumento9 pagineABCD Vocal Technique Handout PDFRanieque RamosNessuna valutazione finora
- Allen and Hightower Population Dynamics Chapter IFM3Documento38 pagineAllen and Hightower Population Dynamics Chapter IFM3Emily HaugNessuna valutazione finora
- Human Genetics Concepts and Applications 11Th Edition Ricki Lewis Test Bank Full Chapter PDFDocumento36 pagineHuman Genetics Concepts and Applications 11Th Edition Ricki Lewis Test Bank Full Chapter PDFdavid.cordero230100% (14)
- Multiple AlleleDocumento3 pagineMultiple AlleleAdroit SangmaNessuna valutazione finora
- An Examination and Critique of Current Methods To Determine Exercise IntensityDocumento28 pagineAn Examination and Critique of Current Methods To Determine Exercise IntensityPabloAñonNessuna valutazione finora
- Biosensors For Environmental ApplicationsDocumento15 pagineBiosensors For Environmental ApplicationsBenjamin HonorioNessuna valutazione finora
- Short Note Biology Form 5-Chapter 3 Coordination and ResponseDocumento6 pagineShort Note Biology Form 5-Chapter 3 Coordination and Responsesalamah_sabri75% (4)
- Chest TubesDocumento34 pagineChest TubesMuhd ShafiqNessuna valutazione finora
- (Springer Series in Wood Science) W. E. Hillis (auth.), John W. Rowe (eds.)-Natural Products of Woody Plants_ Chemicals Extraneous to the Lignocellulosic Cell Wall-Springer-Verlag Berlin Heidelberg (1.pdfDocumento1.274 pagine(Springer Series in Wood Science) W. E. Hillis (auth.), John W. Rowe (eds.)-Natural Products of Woody Plants_ Chemicals Extraneous to the Lignocellulosic Cell Wall-Springer-Verlag Berlin Heidelberg (1.pdfArmandoNessuna valutazione finora
- 7 Pearson SquareDocumento19 pagine7 Pearson Squareapi-235944649Nessuna valutazione finora
- Skin and Body Membranes: EssentialsDocumento43 pagineSkin and Body Membranes: EssentialsJohn Rex Lloyd TunaNessuna valutazione finora
- Experiment 8-Genotypic VariationDocumento3 pagineExperiment 8-Genotypic VariationNUR SABRINA MOHD SHAHNessuna valutazione finora
- Sri Roth 2000Documento11 pagineSri Roth 2000ottoojuniiorNessuna valutazione finora
- Gen. Zoo. Final ReviewerDocumento34 pagineGen. Zoo. Final ReviewerAshley FranciscoNessuna valutazione finora
- Test On Gmos GM Foods Cloning b1b2 Grammar Drills Information Gap Activities Reading 96207Documento4 pagineTest On Gmos GM Foods Cloning b1b2 Grammar Drills Information Gap Activities Reading 96207Thu Quynh NguyenNessuna valutazione finora
- Form 2 Notes-1Documento53 pagineForm 2 Notes-1William NjorogeNessuna valutazione finora
- 300 One Word Substitutions Asked in SSC IBPS UPSC ExamDocumento12 pagine300 One Word Substitutions Asked in SSC IBPS UPSC ExamRashid AliNessuna valutazione finora
- A New Species of Riverine Crab of The Genus Sundathelphusa Bott, 1969 (Crustacea: Brachyura: Gecarcinucidae) From Northeastern Luzon, PhilippinesDocumento10 pagineA New Species of Riverine Crab of The Genus Sundathelphusa Bott, 1969 (Crustacea: Brachyura: Gecarcinucidae) From Northeastern Luzon, PhilippinesNel YagosNessuna valutazione finora
- CV Christopher Lean Jan 2024 Without ReferencesDocumento5 pagineCV Christopher Lean Jan 2024 Without Referencesapi-399765628Nessuna valutazione finora
- Killing of Kangaroos For Meat and SkinDocumento34 pagineKilling of Kangaroos For Meat and SkinVegan FutureNessuna valutazione finora
- The Effect of Temperature On The Reaction Time of The Enzyme Peroxidase - Lab ReportDocumento5 pagineThe Effect of Temperature On The Reaction Time of The Enzyme Peroxidase - Lab ReportAnonymous aZEERFhvNessuna valutazione finora