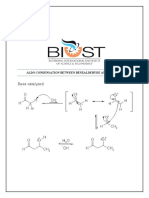
Potrebbero piacerti anche
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesDa EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesNessuna valutazione finora
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeDa EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeNessuna valutazione finora
- FFR 4 PDFDocumento7 pagineFFR 4 PDFCamille Loscalzo100% (1)
- Pre LabghvyicDocumento6 paginePre LabghvyicGobe JamNessuna valutazione finora
- Chem 305 Lab OneDocumento6 pagineChem 305 Lab OneGobe JamNessuna valutazione finora
- Effect of Poly (Acrylic Acid) End-Group Functionality On Inhibition of Calcium Oxalate Crystal GrowthDocumento7 pagineEffect of Poly (Acrylic Acid) End-Group Functionality On Inhibition of Calcium Oxalate Crystal GrowthPencils SharpenerNessuna valutazione finora
- Chalcone: A Versatile Molecule: IntroductionDocumento12 pagineChalcone: A Versatile Molecule: IntroductionKavita TyagiNessuna valutazione finora
- Synthesis of 2-Acetylcyclohexanone Using Pyrrolidine-EnamineDocumento3 pagineSynthesis of 2-Acetylcyclohexanone Using Pyrrolidine-Enaminerobet12Nessuna valutazione finora
- DehydrationDocumento19 pagineDehydrationapi-338215029Nessuna valutazione finora
- Synthesis of Cyclohexanol To Cyclohexene - Lab ReportDocumento5 pagineSynthesis of Cyclohexanol To Cyclohexene - Lab ReportparisdelapenaNessuna valutazione finora
- CHEM F110 - Lab Manual - Nov 5-2020Documento45 pagineCHEM F110 - Lab Manual - Nov 5-2020STUTI MATHUR100% (2)
- Aldol Condensation Reaction PDFDocumento6 pagineAldol Condensation Reaction PDFaizatNessuna valutazione finora
- Laboratory ReportDocumento11 pagineLaboratory ReportElsayed Refaat Aly MareyNessuna valutazione finora
- OrgochemsampleworkDocumento10 pagineOrgochemsampleworkMakcaNessuna valutazione finora
- Experiment #1Documento7 pagineExperiment #1Lakani Tindiwi YangalaNessuna valutazione finora
- Jurnal EugenolDocumento9 pagineJurnal EugenolMillenia AgustinaNessuna valutazione finora
- Appendix Attempts Towards Synthesis of IbuprofenDocumento12 pagineAppendix Attempts Towards Synthesis of IbuprofenJayNessuna valutazione finora
- Katch UmbelliferoneffrDocumento9 pagineKatch Umbelliferoneffrapi-456902531Nessuna valutazione finora
- CyclohexeneDocumento11 pagineCyclohexeneanon-407590100% (10)
- (R) - 3-Hydroxybutanoic Acid Methyl Ester Butanoic Acid, 3-Hydroxy-, (R) - Butanoic Acid, 3-Hydroxy-, Methyl Ester, (R)Documento6 pagine(R) - 3-Hydroxybutanoic Acid Methyl Ester Butanoic Acid, 3-Hydroxy-, (R) - Butanoic Acid, 3-Hydroxy-, Methyl Ester, (R)ArifSheriffNessuna valutazione finora
- Synthesis Report 41 Part 1Documento8 pagineSynthesis Report 41 Part 1tubagussinggihNessuna valutazione finora
- Synthesis of Acetophenone DerivativesDocumento6 pagineSynthesis of Acetophenone DerivativesAwad SaidNessuna valutazione finora
- Inorganic Chemistry Volume 50 Issue 20 2011Documento12 pagineInorganic Chemistry Volume 50 Issue 20 2011Lee ToulouseNessuna valutazione finora
- Expt6 Synthesis of An Alkyl Halide DraftDocumento6 pagineExpt6 Synthesis of An Alkyl Halide DraftAnna Sophia EbuenNessuna valutazione finora
- Wacker Oxidation: A. General Description of The ReactionDocumento6 pagineWacker Oxidation: A. General Description of The Reactionnabila OktavianiNessuna valutazione finora
- Chm557 Laboratory Report: Experiment 4 The Aldol Condensation Reaction: Preparation of DibenzalacetoneDocumento17 pagineChm557 Laboratory Report: Experiment 4 The Aldol Condensation Reaction: Preparation of DibenzalacetonesyafNessuna valutazione finora
- Alquilacion de Arenos Con AlcoholesDocumento5 pagineAlquilacion de Arenos Con AlcoholesJosé Guadalupe García EstradaNessuna valutazione finora
- Articulo 10.-Boric Acid As A Green Catalyst For The Conversion of Aldehydes and KetonesDocumento8 pagineArticulo 10.-Boric Acid As A Green Catalyst For The Conversion of Aldehydes and KetonesPaul Delgado MendozaNessuna valutazione finora
- Preparation of Cyclohexene From CyclohexanolDocumento7 paginePreparation of Cyclohexene From CyclohexanolDumile Nombasa100% (5)
- Experimen 5 Organic ChemistryDocumento8 pagineExperimen 5 Organic ChemistryAbd RaHmanNessuna valutazione finora
- Analysis of Elimination Reaction of CyclohexanolDocumento4 pagineAnalysis of Elimination Reaction of CyclohexanolPratiwi Surya RahayuNessuna valutazione finora
- Working With Hazardous Chemicals: A Publication of Reliable Methods For The Preparation of Organic CompoundsDocumento6 pagineWorking With Hazardous Chemicals: A Publication of Reliable Methods For The Preparation of Organic Compoundsrajesh kothariNessuna valutazione finora
- Aldol Condensation 1Documento8 pagineAldol Condensation 1Bilal100% (2)
- (2S) - ( ) - 3-Exo - (DIMETHYLAMINO) ISOBORNEOL ( (2S) - ( ) - DAIB)Documento6 pagine(2S) - ( ) - 3-Exo - (DIMETHYLAMINO) ISOBORNEOL ( (2S) - ( ) - DAIB)Alex CumbaNessuna valutazione finora
- Catalysis Communications: Suresh D. Salim, Krishnacharya G. AkamanchiDocumento4 pagineCatalysis Communications: Suresh D. Salim, Krishnacharya G. AkamanchinileshsalunkheNessuna valutazione finora
- Cyclohexanol DehydrationDocumento4 pagineCyclohexanol DehydrationVersiformNessuna valutazione finora
- Oxidation of Cyclohexane and Ethylbenzene by Hydrogen Peroxide Over Co-Substituted Heteropolytungstate CatalystDocumento6 pagineOxidation of Cyclohexane and Ethylbenzene by Hydrogen Peroxide Over Co-Substituted Heteropolytungstate Catalystrungrawin ngamkhumNessuna valutazione finora
- Preparation of A-Perfluoroalkyl Ketones From Ab-UnDocumento5 paginePreparation of A-Perfluoroalkyl Ketones From Ab-UnnkazemiNessuna valutazione finora
- New Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsDocumento6 pagineNew Montmorillonite Silylpropylethylenediamine Palladium (II) Complex in Oxidation of Terminal OlefinsChamula K MasNessuna valutazione finora
- A Simple, Green and One-Pot Four-Component Synthesis of 1,4-Dihydropyridines and Their AromatizationDocumento6 pagineA Simple, Green and One-Pot Four-Component Synthesis of 1,4-Dihydropyridines and Their AromatizationEdgar HernándezNessuna valutazione finora
- Methyl SalicylateDocumento10 pagineMethyl Salicylatekab56067% (3)
- A Practical Synthesis of (-) - Kainic AcidDocumento11 pagineA Practical Synthesis of (-) - Kainic AcidNikhil SarpateNessuna valutazione finora
- Studies On ClaisenDocumento14 pagineStudies On ClaisenU4x DanteNessuna valutazione finora
- 2 Functional Group Conversion - 2004 - Advanced Free Radical Reactions For Organic SynthesisDocumento18 pagine2 Functional Group Conversion - 2004 - Advanced Free Radical Reactions For Organic SynthesisShuai LiuNessuna valutazione finora
- Formal Report For Synthesis of An Alkyl HalideDocumento5 pagineFormal Report For Synthesis of An Alkyl HalideLovelyn Marie Morada Nievales80% (5)
- Preparation of Butyl Acetate PDFDocumento6 paginePreparation of Butyl Acetate PDFjoiya100133% (3)
- Synthesis and Characterization of DibenzalacetoneDocumento7 pagineSynthesis and Characterization of DibenzalacetoneTan Yong Jie100% (7)
- Esterification Process To Synthesize Isopropyl Chloroacetate Catalyzed by Lanthanum Dodecyl SulfateDocumento6 pagineEsterification Process To Synthesize Isopropyl Chloroacetate Catalyzed by Lanthanum Dodecyl SulfateVinay JainNessuna valutazione finora
- Radical Coupling ReactionDocumento7 pagineRadical Coupling ReactionGobe JamNessuna valutazione finora
- Acetaldehyde 16.05.2020Documento3 pagineAcetaldehyde 16.05.2020sathiya sathiyaNessuna valutazione finora
- Aldol Condensation LabDocumento5 pagineAldol Condensation Labnmc515288% (8)
- Acetaldehyde Report - Final PDFDocumento20 pagineAcetaldehyde Report - Final PDFDinesh guhanNessuna valutazione finora
- Α-Allenic Esters From Α-Phosphoranylidene Esters And Acid ChloridesDocumento5 pagineΑ-Allenic Esters From Α-Phosphoranylidene Esters And Acid ChloridesJarrett RobinsonNessuna valutazione finora
- Exp 3-Reduction of Cyclohexanone With Sodium BorohydrideDocumento11 pagineExp 3-Reduction of Cyclohexanone With Sodium Borohydrideakuserai100% (3)
- Octyl AcetateDocumento6 pagineOctyl AcetateKristine BautistaNessuna valutazione finora
- Synthesis of Tert-Butyl Chloride Through Hydrochlorination of Tert-Butyl Alcohol and Purification Using DistillationDocumento9 pagineSynthesis of Tert-Butyl Chloride Through Hydrochlorination of Tert-Butyl Alcohol and Purification Using DistillationAnonymous GO6JVW9Wud100% (2)
- Chem 31.1 FR StuffDocumento4 pagineChem 31.1 FR StuffKazaTsuki100% (1)
- Etherification ReportDocumento7 pagineEtherification ReportEwout KesselsNessuna valutazione finora
- Benzylidene AcetalDocumento9 pagineBenzylidene AcetalsadiaNessuna valutazione finora
- Advanced Pharmaceutical analysisDa EverandAdvanced Pharmaceutical analysisValutazione: 4.5 su 5 stelle4.5/5 (2)
- 123.312 Advanced Organic Chemistry: Retrosynthesis: TutorialDocumento10 pagine123.312 Advanced Organic Chemistry: Retrosynthesis: TutorialĐàoTrungHiếuNessuna valutazione finora
- Lab Activity 5Documento5 pagineLab Activity 5Jasmin CeciliaNessuna valutazione finora
- Structural IsomerismDocumento17 pagineStructural IsomerismMotivational BabaNessuna valutazione finora
- Bacterial Phenylalanine and Phenylacetate Catabolic Pathway RevealedDocumento6 pagineBacterial Phenylalanine and Phenylacetate Catabolic Pathway RevealedKarla KetiNessuna valutazione finora
- Aldol ConclusionDocumento1 paginaAldol Conclusionapi-235187189100% (1)
- Solutions - AIATS Medical-2019 (XII Studying&Passed) - Mock Test-4 - (Code-A & B) - (28-04-2019) PDFDocumento36 pagineSolutions - AIATS Medical-2019 (XII Studying&Passed) - Mock Test-4 - (Code-A & B) - (28-04-2019) PDFHarshNessuna valutazione finora
- Carbonyl Compounds 230Documento60 pagineCarbonyl Compounds 230mohtasim hasanNessuna valutazione finora
- Acs Jchemed 5b00170Documento5 pagineAcs Jchemed 5b00170Aitor PastorNessuna valutazione finora
- CHEM 2425. Chapter 22. Carbonyl Alpha-Substitution Reactions (Homework) WDocumento17 pagineCHEM 2425. Chapter 22. Carbonyl Alpha-Substitution Reactions (Homework) WPhương NguyễnNessuna valutazione finora
- (Lecture 3) Carbonyls and AminesDocumento34 pagine(Lecture 3) Carbonyls and AminesKasraSrNessuna valutazione finora
- Green Adeptness in The Synthesis and Stabilization of Copper Nanoparticles: Catalytic, Antibacterial, Cytotoxicity, and Antioxidant ActivitiesDocumento15 pagineGreen Adeptness in The Synthesis and Stabilization of Copper Nanoparticles: Catalytic, Antibacterial, Cytotoxicity, and Antioxidant ActivitiesWindah AnisyahNessuna valutazione finora
- Aldehydes and Ketones FinalDocumento67 pagineAldehydes and Ketones FinalAnil Kumar VermaNessuna valutazione finora
- Keto-Enol Tautomerism - WikipediaDocumento4 pagineKeto-Enol Tautomerism - WikipediafaustoNessuna valutazione finora
- Reaction Mechanism: C C H H H O Hgso H SO CHO H CDocumento6 pagineReaction Mechanism: C C H H H O Hgso H SO CHO H CFATHIMA THANHA T NNessuna valutazione finora
- Organic SynthesisDocumento35 pagineOrganic SynthesisAshok PareekNessuna valutazione finora
- CarboanionDocumento23 pagineCarboanionrajendraNessuna valutazione finora
- Part-1 Stereochemistry of Organic CompoundsDocumento28 paginePart-1 Stereochemistry of Organic CompoundsIct Pfa ClubNessuna valutazione finora
- Chapter 9 Lecture PDFDocumento128 pagineChapter 9 Lecture PDFjoseph changNessuna valutazione finora
- Carbon-Carbon Bond FormationDocumento18 pagineCarbon-Carbon Bond FormationFaTin AziEyatiNessuna valutazione finora
- Vinylic Carbon, Ambient Nucleophilic and Substrate ReactionDocumento11 pagineVinylic Carbon, Ambient Nucleophilic and Substrate Reactiongmanooj_484213209Nessuna valutazione finora
- Enol Dan EnolatDocumento40 pagineEnol Dan EnolatRiyan KateeNessuna valutazione finora
- Organic ChemDocumento116 pagineOrganic ChemNidhi Sisodia100% (1)
- Organic Chemistry ACS Sample QuestionsDocumento20 pagineOrganic Chemistry ACS Sample QuestionsNajmusawwa Aulia RahmahNessuna valutazione finora
- Reaction Mechanism IDocumento15 pagineReaction Mechanism IFilmodeNessuna valutazione finora
- Stereo ChemistryDocumento45 pagineStereo Chemistrysanjanaganguly2005Nessuna valutazione finora
- Heterocyclic ChemistryDocumento57 pagineHeterocyclic ChemistryGuillaume WNessuna valutazione finora
- Selective Hydrogenation of Phenol: Chemnanomat February 2018Documento21 pagineSelective Hydrogenation of Phenol: Chemnanomat February 2018Deo TolloNessuna valutazione finora
- MCAT Organic ChemistryDocumento7 pagineMCAT Organic ChemistryjoNessuna valutazione finora
- Lab 1 Dehydrogenation HydrogenationDocumento3 pagineLab 1 Dehydrogenation HydrogenationJason Chen100% (1)
- DPP 01Documento3 pagineDPP 01Rajeev GangwarNessuna valutazione finora