Potrebbero piacerti anche
- THE EFFECT OF LIGHT INTENSITY ON THE STABILITY OF THE CARR-PRICE REACTION FOR VITAMIN A DETERMINATIONDocumento7 pagineTHE EFFECT OF LIGHT INTENSITY ON THE STABILITY OF THE CARR-PRICE REACTION FOR VITAMIN A DETERMINATIONros4_swe3ty4386Nessuna valutazione finora
- Ed 050 P 626Documento3 pagineEd 050 P 626laurrensiaNessuna valutazione finora
- Separation and Identification of Myoglobin by Paper Chromatography and Protein Assay by Bradford MethodDocumento6 pagineSeparation and Identification of Myoglobin by Paper Chromatography and Protein Assay by Bradford MethodJason Montesa100% (1)
- Electrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsDa EverandElectrochemistry of Dihydroxybenzene Compounds: Carbon Based Electrodes and Their Uses in Synthesis and SensorsNessuna valutazione finora
- Validation of Analysis Method For Determining Content of Chlorpheniramine Malate (CTM) in Tablet Preparation by Uv SpectrophotometryDocumento6 pagineValidation of Analysis Method For Determining Content of Chlorpheniramine Malate (CTM) in Tablet Preparation by Uv SpectrophotometryZakisyarifudintaqiyNessuna valutazione finora
- Course Code:: PHR-322: Pharmaceutical Analysis-LlDocumento7 pagineCourse Code:: PHR-322: Pharmaceutical Analysis-LlMd.Mahfuzur RahmanNessuna valutazione finora
- An Optimised Method To Determine The Degree of Acetylation of Chitin and Chitosan by FTIR SpectrosDocumento8 pagineAn Optimised Method To Determine The Degree of Acetylation of Chitin and Chitosan by FTIR SpectrosLe Thanh LongNessuna valutazione finora
- Use and Calibration of SpectrophotometerDocumento11 pagineUse and Calibration of SpectrophotometerTinashe W MangwandaNessuna valutazione finora
- Thermo Beer LambertDocumento2 pagineThermo Beer LambertRizka Rinda PramastiNessuna valutazione finora
- A Simple and Validated RP-HPLC Method For The Estimation of Methylcobalamin in Bulk and Capsule Dosage FormDocumento4 pagineA Simple and Validated RP-HPLC Method For The Estimation of Methylcobalamin in Bulk and Capsule Dosage FormLayli AmaliaNessuna valutazione finora
- UNIT: SpectrophotometryDocumento15 pagineUNIT: SpectrophotometrybiddyusmcNessuna valutazione finora
- Rapid Determination of Benzalkonium Chloride in A Cosmetic: Key WordsDocumento4 pagineRapid Determination of Benzalkonium Chloride in A Cosmetic: Key WordsYolby Milena Rodriguez ArizaNessuna valutazione finora
- 11 Chapter 3Documento24 pagine11 Chapter 3Imran KakatiNessuna valutazione finora
- By M. J. Caldwell and D. B. Parrish (From The Kansas Agricultural Experiment Manhattan) (Received For Publication, December 26, 1944)Documento7 pagineBy M. J. Caldwell and D. B. Parrish (From The Kansas Agricultural Experiment Manhattan) (Received For Publication, December 26, 1944)Indah WulansariNessuna valutazione finora
- Photocatalytic Degradation of Rhodamine-B DyeDocumento12 paginePhotocatalytic Degradation of Rhodamine-B DyeAmoluck BhatiaNessuna valutazione finora
- Analisis Simetikon PDFDocumento9 pagineAnalisis Simetikon PDFArini Musfiroh50% (2)
- Cyclic Voltammetric Determination of AcetaminophenDocumento5 pagineCyclic Voltammetric Determination of Acetaminophencaanmaro17Nessuna valutazione finora
- Determination of Free Lime in ClinkerDocumento3 pagineDetermination of Free Lime in Clinkerfaheemqc100% (3)
- Applications of Zeeman Graphite Furnace Atomic Absorption Spectrometry in the Chemical Laboratory and in ToxicologyDa EverandApplications of Zeeman Graphite Furnace Atomic Absorption Spectrometry in the Chemical Laboratory and in ToxicologyC. MinoiaNessuna valutazione finora
- Doehlert I PDFDocumento8 pagineDoehlert I PDFAna María TorresNessuna valutazione finora
- 10.1515 - Revac 2022 0039Documento12 pagine10.1515 - Revac 2022 0039yordanosezerihun07Nessuna valutazione finora
- 04 Test Pharmaceutical ChemistryDocumento115 pagine04 Test Pharmaceutical ChemistryThuongNguyen1981Nessuna valutazione finora
- Atr Ft-Ir Imaging of Acetic AcidDocumento10 pagineAtr Ft-Ir Imaging of Acetic AcidMon RonquilloNessuna valutazione finora
- Spectrophotometric Determination of Isoxsuprine in Pure and Pharmaceutical FormsDocumento7 pagineSpectrophotometric Determination of Isoxsuprine in Pure and Pharmaceutical FormsIOSRjournalNessuna valutazione finora
- Lab Manual Adv Food Analysis-2 - Uv VisDocumento3 pagineLab Manual Adv Food Analysis-2 - Uv VisyusrydejaneNessuna valutazione finora
- Alternatives To The in Do Phenol Blue MethodDocumento15 pagineAlternatives To The in Do Phenol Blue MethodoverspongNessuna valutazione finora
- River Analyzer For Chlorotriazines With A Direct Optical ImmunosensorDocumento7 pagineRiver Analyzer For Chlorotriazines With A Direct Optical ImmunosensorTadeu ViannaNessuna valutazione finora
- E60 Analysis of Metals, Ores, and Related Materials by Molecular Absorption Spectrometry1Documento8 pagineE60 Analysis of Metals, Ores, and Related Materials by Molecular Absorption Spectrometry1Electra AkbarillahNessuna valutazione finora
- 697651Documento10 pagine697651Anonymous JvptVyNsNessuna valutazione finora
- 230778-Article Text-560167-1-10-20220830Documento11 pagine230778-Article Text-560167-1-10-20220830yordanosezerihun07Nessuna valutazione finora
- Thermometric Titrimetry: International Series of Monographs in Analytical ChemistryDa EverandThermometric Titrimetry: International Series of Monographs in Analytical ChemistryNessuna valutazione finora
- BE184P Exercise 2.1 Chromatographic Analysis - HPLCDocumento10 pagineBE184P Exercise 2.1 Chromatographic Analysis - HPLCDen CelestraNessuna valutazione finora
- Oser1945 PDFDocumento4 pagineOser1945 PDFJavier Palermo Maita NoelNessuna valutazione finora
- HPLC Article - 1Documento8 pagineHPLC Article - 1akkimipadmaNessuna valutazione finora
- Determination and Validation of Uv Spectrophotometric Method For Estimation of Bicalutamide TabletDocumento5 pagineDetermination and Validation of Uv Spectrophotometric Method For Estimation of Bicalutamide TabletGembong Van BeethovenNessuna valutazione finora
- DOC042 53 20249 Oct16 PDFDocumento7 pagineDOC042 53 20249 Oct16 PDFLinh ĐỗNessuna valutazione finora
- Full Text Paper-Gas ChromatographyDocumento2 pagineFull Text Paper-Gas ChromatographyRitesh AgarwalNessuna valutazione finora
- CH 304 ANALYTICAL CHEMISTRYDocumento4 pagineCH 304 ANALYTICAL CHEMISTRYMike VhurinosharaNessuna valutazione finora
- Assays MOST IodisedsaltDocumento4 pagineAssays MOST IodisedsaltJasdeep KaurNessuna valutazione finora
- UV Lab Report Yy GroupDocumento14 pagineUV Lab Report Yy GroupFatin Nasuha MahyuddinNessuna valutazione finora
- Bmri2013 545703Documento10 pagineBmri2013 545703Rizkaa Harinii QuinshaNessuna valutazione finora
- HPLC Method For Determinationof Ascorbic Acid in Fruit and VegatablesDocumento8 pagineHPLC Method For Determinationof Ascorbic Acid in Fruit and VegatablesHuong NguyenNessuna valutazione finora
- Chemistry 460 Problems Set 1 Statistics and Experimental DesignDocumento69 pagineChemistry 460 Problems Set 1 Statistics and Experimental Designvanhiepk52a100% (1)
- App Note 323 ThermoDocumento8 pagineApp Note 323 ThermoLoreto VillegasNessuna valutazione finora
- Analytica Chimica Acta: Taher Alizadeh, Mohammad Reza Ganjali, Faride Ra FieiDocumento9 pagineAnalytica Chimica Acta: Taher Alizadeh, Mohammad Reza Ganjali, Faride Ra FieiwardaninurindahNessuna valutazione finora
- 460 Bai Tap Dien HoaDocumento69 pagine460 Bai Tap Dien HoaTiến Thành Nguyễn50% (2)
- Topiramato 1Documento6 pagineTopiramato 1Daniel NicolásNessuna valutazione finora
- Beers Law SpectrophotometryDocumento4 pagineBeers Law SpectrophotometrySandhya KumariNessuna valutazione finora
- ICP-MS ELEMENTAL ANALYSIS OF WATER AND WASTE SAMPLESDocumento18 pagineICP-MS ELEMENTAL ANALYSIS OF WATER AND WASTE SAMPLESMike CoronaNessuna valutazione finora
- S12 1011 The Use of The Spectrophotometer and Beers LawDocumento7 pagineS12 1011 The Use of The Spectrophotometer and Beers LawDr.Santosh KumarNessuna valutazione finora
- (Shimadzu) Appl News No A564 Measurement of Li in Seawater (Flame Micro Sampling Method)Documento2 pagine(Shimadzu) Appl News No A564 Measurement of Li in Seawater (Flame Micro Sampling Method)faridsidikNessuna valutazione finora
- A Comparative Study For The Quantitative Determination of Paracetamol in Tablets Using UVDocumento7 pagineA Comparative Study For The Quantitative Determination of Paracetamol in Tablets Using UVRizqita Atikah SNessuna valutazione finora
- 2011 - UV-Vis Iron (III) - Salicylate Complex PDFDocumento11 pagine2011 - UV-Vis Iron (III) - Salicylate Complex PDFEub Eu33% (3)
- Sequential determination of drugs using flow injection analysisDocumento7 pagineSequential determination of drugs using flow injection analysisNur Aini IktikhafsariNessuna valutazione finora
- Group 10 Composition Lab ReportDocumento7 pagineGroup 10 Composition Lab ReportEthan MuddNessuna valutazione finora
- Festival of Contemporary Science WORKSHOPS 30 January 2010: Analytical Chemistry 1: Chromatography: GC-MS, HPLC and TLCDocumento7 pagineFestival of Contemporary Science WORKSHOPS 30 January 2010: Analytical Chemistry 1: Chromatography: GC-MS, HPLC and TLCAhmad AlbabNessuna valutazione finora
- HPLC-UV or GCDocumento6 pagineHPLC-UV or GCrofikudouNessuna valutazione finora
- EXP 7 Simultaneous Determination of Caffeine and Acetylsalicylic Acid in An Analgesic by Ultraviolet SpectrosDocumento8 pagineEXP 7 Simultaneous Determination of Caffeine and Acetylsalicylic Acid in An Analgesic by Ultraviolet Spectroslebogang100% (3)
- Appendix 3 Sample Lab ReportDocumento8 pagineAppendix 3 Sample Lab ReportXiuQingNessuna valutazione finora
- Carotenoids: Nutrition, Analysis and TechnologyDa EverandCarotenoids: Nutrition, Analysis and TechnologyAgnieszka KaczorNessuna valutazione finora
- Modelling Entrained Flow ReactorDocumento7 pagineModelling Entrained Flow ReactorFernanda GuerreroNessuna valutazione finora
- Advantages and Disadvantages of Composition and Properties of Biomass PDFDocumento21 pagineAdvantages and Disadvantages of Composition and Properties of Biomass PDFNatalia Moreno MorenoNessuna valutazione finora
- Ph.d. Thesis - Ke Qin Klar Til (Entrained Flow Gasifier)Documento187 paginePh.d. Thesis - Ke Qin Klar Til (Entrained Flow Gasifier)Fernanda GuerreroNessuna valutazione finora
- Prediction of The Reid Vapor Pressure of Petroleum Fuels: M. R. Riazi, T. A. Albahri and A. H. AlqattanDocumento2 paginePrediction of The Reid Vapor Pressure of Petroleum Fuels: M. R. Riazi, T. A. Albahri and A. H. AlqattanFernanda GuerreroNessuna valutazione finora
- Simulation AspenDocumento2 pagineSimulation AspenFernanda GuerreroNessuna valutazione finora
- Kids 2Documento1 paginaKids 2Fernanda GuerreroNessuna valutazione finora
- Gasification Systm - SwedenDocumento8 pagineGasification Systm - SwedenFernanda GuerreroNessuna valutazione finora
- Simulation of Biomass GasificationDocumento62 pagineSimulation of Biomass GasificationFernanda Guerrero100% (2)
- JS Activated Economics Plus V84Documento14 pagineJS Activated Economics Plus V84Jessica CehNessuna valutazione finora
- Biomass To Syngas (Gasification)Documento7 pagineBiomass To Syngas (Gasification)Fernanda GuerreroNessuna valutazione finora
- Petrochemical Products ChartDocumento1 paginaPetrochemical Products ChartFernanda GuerreroNessuna valutazione finora
- 2016Documento10 pagine2016Fernanda GuerreroNessuna valutazione finora
- The Use of Hydrogen in Refineries About This ChapterDocumento7 pagineThe Use of Hydrogen in Refineries About This ChapterFernanda GuerreroNessuna valutazione finora
- The Age of Thermal RefineriesDocumento11 pagineThe Age of Thermal RefineriesFernanda GuerreroNessuna valutazione finora
- Articulo SillmanDocumento25 pagineArticulo SillmanFernanda GuerreroNessuna valutazione finora
- RefinacionDocumento4 pagineRefinacionAngelSosa100% (1)
- Simple & Complex RefineriesDocumento6 pagineSimple & Complex RefineriesFernanda Guerrero100% (1)
- Simple & Complex RefineriesDocumento6 pagineSimple & Complex RefineriesFernanda Guerrero100% (1)
- Gasoline Manufacture OverviewDocumento11 pagineGasoline Manufacture OverviewFernanda GuerreroNessuna valutazione finora
- RefiningDocumento4 pagineRefiningAngelSosaNessuna valutazione finora
- Vocabulary For TOEFL iBTDocumento191 pagineVocabulary For TOEFL iBTquevinh94% (48)
- Name:: A. Complete The Following Sentences With The Past of Such Verb. B. Name Each Mean of TransportationDocumento1 paginaName:: A. Complete The Following Sentences With The Past of Such Verb. B. Name Each Mean of TransportationFernanda GuerreroNessuna valutazione finora
- Pronunciation of Regular Past Tense Verbs WorksheetDocumento2 paginePronunciation of Regular Past Tense Verbs WorksheetJorge Iván Albear GuevaraNessuna valutazione finora
- Scilab codes for chemical reaction engineeringDocumento105 pagineScilab codes for chemical reaction engineeringyvehuangNessuna valutazione finora
- UNIT CONVERSION FACTORSDocumento2 pagineUNIT CONVERSION FACTORSChemistNessuna valutazione finora
- Fluidos de PerforacionDocumento0 pagineFluidos de PerforacionFernanda GuerreroNessuna valutazione finora
- Visual Basic TutorialDocumento21 pagineVisual Basic TutorialUmesh Prasad100% (1)
- Elementarygrammargames SplitDocumento3 pagineElementarygrammargames SplitFernanda GuerreroNessuna valutazione finora
- Sbl-Report3 ProteinDocumento8 pagineSbl-Report3 Proteinapi-383715002Nessuna valutazione finora
- YS3060 Spectrophotometer Operating Manual - 20170515 PDFDocumento47 pagineYS3060 Spectrophotometer Operating Manual - 20170515 PDFNelson BarriosNessuna valutazione finora
- Experiment Report: Spectrophotometric Analysis of Caffeine and Benzoic Acid in Soft DrinkDocumento12 pagineExperiment Report: Spectrophotometric Analysis of Caffeine and Benzoic Acid in Soft DrinkNitty MeYaNessuna valutazione finora
- PEGylated RosinDocumento9 paginePEGylated RosindineshmorkhadeNessuna valutazione finora
- Sunflower Oil Bleaching by Adsorption Onto Acid-Activated BentoniteDocumento6 pagineSunflower Oil Bleaching by Adsorption Onto Acid-Activated BentoniteramcatNessuna valutazione finora
- Model 6305 UV/Visible Range Spectrophotometer Operating ManualDocumento19 pagineModel 6305 UV/Visible Range Spectrophotometer Operating Manualhendreen ibrahimNessuna valutazione finora
- Shimadzu CuvetteGuide 2014Documento27 pagineShimadzu CuvetteGuide 2014Elizabeth CarrNessuna valutazione finora
- Beers Law SpectrophotometryDocumento4 pagineBeers Law SpectrophotometrySandhya KumariNessuna valutazione finora
- Operation and Calibration of UV-VIS SpectrophotometerDocumento8 pagineOperation and Calibration of UV-VIS SpectrophotometerMaruthi K100% (1)
- Manual Analysis Methods For The Brewery Industry Prove 05 2021 Final WebDocumento130 pagineManual Analysis Methods For The Brewery Industry Prove 05 2021 Final WebleidyNessuna valutazione finora
- Test 2014Documento10 pagineTest 2014sahseatranNessuna valutazione finora
- Viscera PreservationDocumento15 pagineViscera Preservationniraj_sdNessuna valutazione finora
- UV Visible Spectrophotometric Method of Paracetamol Tablet FormulationDocumento5 pagineUV Visible Spectrophotometric Method of Paracetamol Tablet FormulationGabriel GabiNessuna valutazione finora
- Spectrophotometric Determination of Iron in Aqueous Solutions As A Complex of 1,10-PhenanthrolineDocumento4 pagineSpectrophotometric Determination of Iron in Aqueous Solutions As A Complex of 1,10-PhenanthrolineJaimie LojaNessuna valutazione finora
- APHA 23rd Edition ChlorophyllDocumento3 pagineAPHA 23rd Edition ChlorophyllMoutaz AdelNessuna valutazione finora
- Beta - Carotene ExtractionDocumento9 pagineBeta - Carotene ExtractionVyshali PingleNessuna valutazione finora
- Comparative Analysis of Transmittance For Different Types of Commercially Available Zirconia and Lithium Disilicate MaterialsDocumento6 pagineComparative Analysis of Transmittance For Different Types of Commercially Available Zirconia and Lithium Disilicate MaterialsNaoki MezarinaNessuna valutazione finora
- Priyank Patoliya: BE Biomedical EngineeringDocumento3 paginePriyank Patoliya: BE Biomedical EngineeringKirtan KananiNessuna valutazione finora
- Instrumentation What Is Istrumentation?Documento5 pagineInstrumentation What Is Istrumentation?lintasamimNessuna valutazione finora
- Industrial - PDF p03715Documento103 pagineIndustrial - PDF p03715Dyeing DyeingNessuna valutazione finora
- Color Determination of Plastic Pellets: Standard Test Method ForDocumento5 pagineColor Determination of Plastic Pellets: Standard Test Method ForSudarshan Daw100% (1)
- Spectrophotometric Determination of The Acid Dissociation Constant of Methyl RedDocumento3 pagineSpectrophotometric Determination of The Acid Dissociation Constant of Methyl Red7063673nasNessuna valutazione finora
- Spectral Analysis System UniSpec-SCDocumento4 pagineSpectral Analysis System UniSpec-SCJuan Galindo Muñiz0% (1)
- Practical 1 - Spectrophotometry TechniquesDocumento13 paginePractical 1 - Spectrophotometry TechniquesDhanen DranNessuna valutazione finora
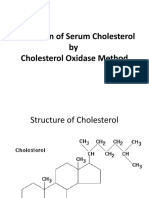
- Estimation of Serum CholesterolDocumento33 pagineEstimation of Serum CholesterolNihalNessuna valutazione finora
- HunterLab Brochure For Plastic IndustryDocumento4 pagineHunterLab Brochure For Plastic IndustryJohnny LukeNessuna valutazione finora
- Two-component spectrophotometric analysis methodDocumento4 pagineTwo-component spectrophotometric analysis methodiabureid7460Nessuna valutazione finora
- Antioxidantes en ComprimidosDocumento12 pagineAntioxidantes en ComprimidosJose Andres RodriguezNessuna valutazione finora
- Propagation Loss in Optical FibersDocumento24 paginePropagation Loss in Optical FibersEr Arvind Kumar MehraNessuna valutazione finora
- Astm D1607-91 (2018)Documento5 pagineAstm D1607-91 (2018)Carlos SDNessuna valutazione finora