Potrebbero piacerti anche
- Bi 00444 A 006Documento8 pagineBi 00444 A 006Moh SyaifudinNessuna valutazione finora
- tmp80B0 TMPDocumento24 paginetmp80B0 TMPFrontiersNessuna valutazione finora
- 1 s2.0 S1090780701924991 MainDocumento13 pagine1 s2.0 S1090780701924991 MainJoão VictorNessuna valutazione finora
- Solvent Effects On The Singlet - Triplet Equilibrium and Reactivity of A Ground Triplet State Arylalkyl CarbeneDocumento4 pagineSolvent Effects On The Singlet - Triplet Equilibrium and Reactivity of A Ground Triplet State Arylalkyl CarbeneSergioSilvaNessuna valutazione finora
- Zacharias 2002 Science - Partitioning of LipidDocumento4 pagineZacharias 2002 Science - Partitioning of LipidAlfun IqbalNessuna valutazione finora
- Biophysical Characteristics The Replication Arrest ProteinDocumento8 pagineBiophysical Characteristics The Replication Arrest Proteinrfahad22926Nessuna valutazione finora
- Structural and Functional Consequences of Altering A Peptide MHC Anchor ResidueDocumento10 pagineStructural and Functional Consequences of Altering A Peptide MHC Anchor ResidueWilliam AgudeloNessuna valutazione finora
- Macromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976Da EverandMacromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976B. SedláčekNessuna valutazione finora
- Design and Synthesis of Single-Nanoparticle Optical Biosensors For Imaging and Characterization of Single Receptor Molecules On Single Living CellsDocumento11 pagineDesign and Synthesis of Single-Nanoparticle Optical Biosensors For Imaging and Characterization of Single Receptor Molecules On Single Living Cellsthuang666Nessuna valutazione finora
- Probing The Interaction Between Two Single Molecules: Fluorescence Resonance Energy Transfer Between A Single Donor and A Single AcceptorDocumento5 pagineProbing The Interaction Between Two Single Molecules: Fluorescence Resonance Energy Transfer Between A Single Donor and A Single AcceptorMelbaNeliaEspinosaNessuna valutazione finora
- Synthesis and DFT Studies of Novel Aryloxymaleimides Via Nucleophilic Substitution of Tosyloxy GroupDocumento5 pagineSynthesis and DFT Studies of Novel Aryloxymaleimides Via Nucleophilic Substitution of Tosyloxy GroupCINDY VANESSA RESTREPO BURGOSNessuna valutazione finora
- Dissertation NMRDocumento5 pagineDissertation NMRFindSomeoneToWriteMyCollegePaperUK100% (1)
- References and Notes: Soc., NDocumento3 pagineReferences and Notes: Soc., NMohammed ZiyadNessuna valutazione finora
- N Comms 4999Documento6 pagineN Comms 4999Blake KellyNessuna valutazione finora
- Tsumoto 1994Documento6 pagineTsumoto 1994j.franco0483Nessuna valutazione finora
- Vrouw, Mar 2011Documento4 pagineVrouw, Mar 2011emediageNessuna valutazione finora
- Modification of Photochemical Reactivity by Zeolites: Cation Controlled Photodimerisation of Acenaphthylene Within FaujasitesDocumento4 pagineModification of Photochemical Reactivity by Zeolites: Cation Controlled Photodimerisation of Acenaphthylene Within FaujasitesTrịnh Xuân LộcNessuna valutazione finora
- Simulation of Positron and Electron Elastic Mean Free Path and Diffusion AngleDocumento3 pagineSimulation of Positron and Electron Elastic Mean Free Path and Diffusion Anglejojo sosoNessuna valutazione finora
- Solenoid and Time Projection Chamber For Neutron Lifetime Measurement - LinaDocumento5 pagineSolenoid and Time Projection Chamber For Neutron Lifetime Measurement - LinaLuz PeñaNessuna valutazione finora
- Molecular basis of small-molecule binding to α-synucleinDocumento27 pagineMolecular basis of small-molecule binding to α-synucleinSebastian KmiecikNessuna valutazione finora
- Triplet AND AND: Competitive Energy Electron Transfer Reactions With Trans-Stilbene A, ADocumento5 pagineTriplet AND AND: Competitive Energy Electron Transfer Reactions With Trans-Stilbene A, AStuteeNessuna valutazione finora
- Take A Break, Jul 2011Documento5 pagineTake A Break, Jul 2011emediageNessuna valutazione finora
- Esr Spectra of Oganic Free RadicalDocumento47 pagineEsr Spectra of Oganic Free RadicalAditya MahakalNessuna valutazione finora
- Isabel K. Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu Protein-DNA ComplexDocumento61 pagineIsabel K. Darcy, John Luecke and Mariel Vazquez - Tangle Analysis of Difference Topology Experiments: Applications To A Mu Protein-DNA ComplexLokosooNessuna valutazione finora
- Protein-DNA Interaction EnergeticsDocumento4 pagineProtein-DNA Interaction Energeticsph132Nessuna valutazione finora
- ScienceDocumento13 pagineScienceJosh Go GoNessuna valutazione finora
- Lab Report - Thermodynamics and Kinetics of A Substitution Reaction of A Metal ComplexDocumento16 pagineLab Report - Thermodynamics and Kinetics of A Substitution Reaction of A Metal ComplexValerie MangasarNessuna valutazione finora
- 2011-APL-triplet For OLED - AdachiDocumento4 pagine2011-APL-triplet For OLED - AdachiAftab FarrukhNessuna valutazione finora
- Molecular Dynamics Simulations of The Unfolding of Mutant Prion Variants at Elevated TemperaturesDocumento10 pagineMolecular Dynamics Simulations of The Unfolding of Mutant Prion Variants at Elevated TemperaturesLeslieTettehNessuna valutazione finora
- Quantum Dot Fluorescence Characterizes The Nanoscale Organization of T Cell Receptors For AntigenDocumento3 pagineQuantum Dot Fluorescence Characterizes The Nanoscale Organization of T Cell Receptors For AntigenAditya Budi FauziNessuna valutazione finora
- Inducible Expression of Claudin-1-Myc But Not occludin-VSV-G Results in Aberrant Tight Junction Strand Formation in MDCK CellsDocumento12 pagineInducible Expression of Claudin-1-Myc But Not occludin-VSV-G Results in Aberrant Tight Junction Strand Formation in MDCK CellsKarina B Hernandez ANessuna valutazione finora
- Dynamic Quenching of The Dual Fluorescence of Molecules: Condensed-Matter SpectrosDocumento7 pagineDynamic Quenching of The Dual Fluorescence of Molecules: Condensed-Matter Spectrosprakush_prakushNessuna valutazione finora
- Angela Seidl and Hans-Jurgen Hinz - The Free Energy of DNA Supercoiling Is Enthalpy-DeterminedDocumento5 pagineAngela Seidl and Hans-Jurgen Hinz - The Free Energy of DNA Supercoiling Is Enthalpy-DeterminedDopameNessuna valutazione finora
- Specs Pctru2020050024Documento53 pagineSpecs Pctru2020050024MichaelandKaye DanaoNessuna valutazione finora
- Curves: (Oscl (Co) (Pph3) 2si (Oh) 2) 20Documento2 pagineCurves: (Oscl (Co) (Pph3) 2si (Oh) 2) 20Lazar AlinaNessuna valutazione finora
- 371812b0Documento1 pagina371812b0ViorelDiorducNessuna valutazione finora
- Correlation Between The Glass Transition Temperatures and Repeating Unit Structure For High Molecular Weight PolymersDocumento8 pagineCorrelation Between The Glass Transition Temperatures and Repeating Unit Structure For High Molecular Weight PolymersSinisa Gale GacicNessuna valutazione finora
- Energy landscape of prion protein helix 1Documento10 pagineEnergy landscape of prion protein helix 1Venkata Suryanarayana GorleNessuna valutazione finora
- Theory and Application Voltammetry Measurement of Electrode Reaction KineticsDocumento5 pagineTheory and Application Voltammetry Measurement of Electrode Reaction KineticsJubin KumarNessuna valutazione finora
- Ras - Pandey@Usm - Edu: Thermal Denaturation of A Protein (Cove) by A Coarse-Grained Monte Carlo SimulationDocumento9 pagineRas - Pandey@Usm - Edu: Thermal Denaturation of A Protein (Cove) by A Coarse-Grained Monte Carlo SimulationTiara Ayu F HNessuna valutazione finora
- (1988) Comparison Between DNA Melting Thermodynamics and DNA Polymerase FidelityDocumento5 pagine(1988) Comparison Between DNA Melting Thermodynamics and DNA Polymerase Fidelitydominguezmariela465Nessuna valutazione finora
- Anti-Hidrogen SpectreDocumento12 pagineAnti-Hidrogen SpectreEdgar CuellarNessuna valutazione finora
- Barth, 2007Documento29 pagineBarth, 2007Marco Aurelio OliveiraNessuna valutazione finora
- Theory of Fluorescence Correlation Spectroscopy On Single Molecules, JPhyChemA 2000Documento6 pagineTheory of Fluorescence Correlation Spectroscopy On Single Molecules, JPhyChemA 2000Lei_XuNessuna valutazione finora
- Jong Seol Yuk Et Al - Analysis of Protein Interactions On Protein Arrays by A Wavelength Interrogation-Based Surface Plasmon Resonance BiosensorDocumento9 pagineJong Seol Yuk Et Al - Analysis of Protein Interactions On Protein Arrays by A Wavelength Interrogation-Based Surface Plasmon Resonance BiosensorKorezmNessuna valutazione finora
- Stereoelectronic Effects On The Basicity and Nucleophilicity of Phosphites and Phosphates. Ab Initio Molecular Orbital Calculations and The A-EffectDocumento7 pagineStereoelectronic Effects On The Basicity and Nucleophilicity of Phosphites and Phosphates. Ab Initio Molecular Orbital Calculations and The A-EffectvycttorNessuna valutazione finora
- Electroconformational CouplingDocumento4 pagineElectroconformational CouplingDean AstumianNessuna valutazione finora
- Molecular Dynamics Study of Oligonucleotides Containing DifluorotolueneDocumento9 pagineMolecular Dynamics Study of Oligonucleotides Containing DifluorotolueneSveti JeronimNessuna valutazione finora
- بكتريا PDFDocumento45 pagineبكتريا PDFADEL A. ABDNessuna valutazione finora
- Polar PNA Exp PDFDocumento13 paginePolar PNA Exp PDFIgor GornéNessuna valutazione finora
- 2686 PDFDocumento11 pagine2686 PDFmojNessuna valutazione finora
- Gennarelli 1991Documento3 pagineGennarelli 1991Gerardo David GonzalezNessuna valutazione finora
- Anie 201808861Documento5 pagineAnie 201808861Rajdikshit GogoiNessuna valutazione finora
- New Equations For Redoxand Nano-Signal TransductionDocumento4 pagineNew Equations For Redoxand Nano-Signal TransductionMycoLogist4LifeNessuna valutazione finora
- PNAS 2001 Basché 10527 8Documento2 paginePNAS 2001 Basché 10527 8Jazmín Gonzales TovarNessuna valutazione finora
- Anja Dietrich2002Documento21 pagineAnja Dietrich2002OhisverNessuna valutazione finora
- Energy States of MoleculesDocumento12 pagineEnergy States of MoleculesBenjamín Marc Ridgway de SassouNessuna valutazione finora
- Is Tetramethylene An Intermediate?: Figure 1. Contour Plot A DisDocumento2 pagineIs Tetramethylene An Intermediate?: Figure 1. Contour Plot A DisJosé CortésNessuna valutazione finora
- Josh Vura-Weis Et Al - Crossover From Single-Step Tunneling To Multistep Hopping For Molecular Triplet Energy TransferDocumento4 pagineJosh Vura-Weis Et Al - Crossover From Single-Step Tunneling To Multistep Hopping For Molecular Triplet Energy TransferGomsajNessuna valutazione finora
- Gaining ligand selectivity in thyroid hormone receptors via entropyDocumento6 pagineGaining ligand selectivity in thyroid hormone receptors via entropyRicardo S Falavinha JrNessuna valutazione finora
- Encapsulation of Horseradish Peroxidase-Glucose Oxidase (Hrp-Gox) in Silica Aquagel Synthesized From Rice Hull Ash For Enzymatic Reaction of GlucoseDocumento8 pagineEncapsulation of Horseradish Peroxidase-Glucose Oxidase (Hrp-Gox) in Silica Aquagel Synthesized From Rice Hull Ash For Enzymatic Reaction of GlucoseMoh SyaifudinNessuna valutazione finora
- Perhitungan Deskriptor Dengan Melibatkan Anion Garam: Analisis Hubungan Kuantitatif Struktur-Aktivitas Senyawa Antimalaria Turunan 1,10-Fenan TrolinDocumento8 paginePerhitungan Deskriptor Dengan Melibatkan Anion Garam: Analisis Hubungan Kuantitatif Struktur-Aktivitas Senyawa Antimalaria Turunan 1,10-Fenan TrolinMoh SyaifudinNessuna valutazione finora
- Bi 00145 A 004Documento14 pagineBi 00145 A 004Moh SyaifudinNessuna valutazione finora
- Translate HyperChep7Documento4 pagineTranslate HyperChep7Moh SyaifudinNessuna valutazione finora
- JP 9705075Documento13 pagineJP 9705075Moh SyaifudinNessuna valutazione finora
- ChartDocumento3 pagineChartMoh SyaifudinNessuna valutazione finora
- Apply for Astra 1st ProgramDocumento7 pagineApply for Astra 1st ProgramMoh SyaifudinNessuna valutazione finora
- Contribution of Tryptophan Residues The Combining Site of A Monoclonal Anti Dinitrophenyl Spin-Label Antibody?Documento7 pagineContribution of Tryptophan Residues The Combining Site of A Monoclonal Anti Dinitrophenyl Spin-Label Antibody?Moh SyaifudinNessuna valutazione finora
- Ar 010030 MDocumento8 pagineAr 010030 MMoh SyaifudinNessuna valutazione finora
- JP 011048 HDocumento21 pagineJP 011048 HMoh SyaifudinNessuna valutazione finora
- JP 9705075Documento13 pagineJP 9705075Moh SyaifudinNessuna valutazione finora
- J 100475 A 014Documento13 pagineJ 100475 A 014Moh SyaifudinNessuna valutazione finora
- Ja 00771 A 014Documento14 pagineJa 00771 A 014Moh SyaifudinNessuna valutazione finora
- Dynamics of Reactions in Polar Solvents. Semiclassical Trajectory Studies of Electron-Transfer and Proton-Transfer ReactionsDocumento7 pagineDynamics of Reactions in Polar Solvents. Semiclassical Trajectory Studies of Electron-Transfer and Proton-Transfer ReactionsMoh SyaifudinNessuna valutazione finora
- CR 950045 WDocumento24 pagineCR 950045 WMoh SyaifudinNessuna valutazione finora
- J 100475 A 014Documento13 pagineJ 100475 A 014Moh SyaifudinNessuna valutazione finora
- Electrostatic Basis of Structure-Function Correlation in ProteinsDocumento7 pagineElectrostatic Basis of Structure-Function Correlation in ProteinsMoh SyaifudinNessuna valutazione finora
- Perspectives Biochemistry: How Do Serine Proteases Really Work??Documento9 paginePerspectives Biochemistry: How Do Serine Proteases Really Work??Moh SyaifudinNessuna valutazione finora
- 10 F 01 DrugDiscToday2 457 QSAR QSAR3DDocumento11 pagine10 F 01 DrugDiscToday2 457 QSAR QSAR3DMoh SyaifudinNessuna valutazione finora
- VitaminsDocumento95 pagineVitaminscaksonyNessuna valutazione finora
- When The Koel SingsDocumento15 pagineWhen The Koel SingsAlfie Paduga BeguinaNessuna valutazione finora
- 4.1 - Cell Cycle Part 1Documento5 pagine4.1 - Cell Cycle Part 1Deomar Joseph ParadoNessuna valutazione finora
- Physiological Psychology ReviewerDocumento13 paginePhysiological Psychology ReviewerLeopando Rod CyrenzNessuna valutazione finora
- Organizing A TextDocumento3 pagineOrganizing A TextMercedes Jimenez RomanNessuna valutazione finora
- TC QMM 56942Documento120 pagineTC QMM 56942Fernando R EpilNessuna valutazione finora
- Biology ss2Documento2 pagineBiology ss2DanielNessuna valutazione finora
- Growth and DevelopmentDocumento10 pagineGrowth and DevelopmentPiyush DuttaNessuna valutazione finora
- Post Harvest Handling in LitchiDocumento52 paginePost Harvest Handling in LitchiDr Parag B JadhavNessuna valutazione finora
- Theories of Tooth MovementDocumento11 pagineTheories of Tooth MovementAhmedsy Ahmedsy AhmedsyNessuna valutazione finora
- Evolving Knowledge in Framing of Teratogenic Activity Towards Risk PerceptionDocumento13 pagineEvolving Knowledge in Framing of Teratogenic Activity Towards Risk Perceptionsandy candyNessuna valutazione finora
- Natamycin Story - What You Need to KnowDocumento13 pagineNatamycin Story - What You Need to KnowCharles MardiniNessuna valutazione finora
- Field Inspection Techniques Ifad-CaspDocumento17 pagineField Inspection Techniques Ifad-CaspNasiru Kura100% (1)
- Chiro Guidelines WHODocumento51 pagineChiro Guidelines WHOMinh MinhNessuna valutazione finora
- C Specialized GeneralBiology1 Sem1 Clas3 CellsinPlantand-AnimalTissue v2Documento14 pagineC Specialized GeneralBiology1 Sem1 Clas3 CellsinPlantand-AnimalTissue v2ダギア メイ アニアNessuna valutazione finora
- Self Concept Inventory Hand OutDocumento2 pagineSelf Concept Inventory Hand OutHarold LowryNessuna valutazione finora
- Molecular Genetics of Colorectal Cancer - UpToDateDocumento41 pagineMolecular Genetics of Colorectal Cancer - UpToDateToweran ToweraneNessuna valutazione finora
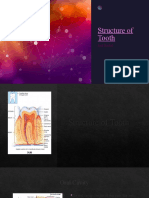
- Structure of Tooth m121Documento13 pagineStructure of Tooth m121amirhossein vaeziNessuna valutazione finora
- Classification and Characterization of Microsatellite Instability Across 18 Cancer TypesDocumento13 pagineClassification and Characterization of Microsatellite Instability Across 18 Cancer TypeshadymatrixNessuna valutazione finora
- Population Size, Density, & Dispersal: Demography: Describing Populations and How They ChangeDocumento4 paginePopulation Size, Density, & Dispersal: Demography: Describing Populations and How They ChangeAllan Jr. Agao-AgaoNessuna valutazione finora
- Module 2 NCM 114Documento11 pagineModule 2 NCM 114Erven AranasNessuna valutazione finora
- Transport Phenomena in Nervous System PDFDocumento538 pagineTransport Phenomena in Nervous System PDFUdayanidhi RNessuna valutazione finora
- Candida Tropicalis - ACMGDocumento12 pagineCandida Tropicalis - ACMGSergio Iván López LallanaNessuna valutazione finora
- WCH03 01 Que 20180124Documento16 pagineWCH03 01 Que 20180124Rameez Mazhar SiddiqiNessuna valutazione finora
- Pecutan Akhir Science 2021Documento29 paginePecutan Akhir Science 2021Azween SabtuNessuna valutazione finora
- Full Download Human Physiology From Cells To Systems 8th Edition Lauralee Sherwood Solutions ManualDocumento36 pagineFull Download Human Physiology From Cells To Systems 8th Edition Lauralee Sherwood Solutions Manualgambolrapinous.ggqcdr100% (39)
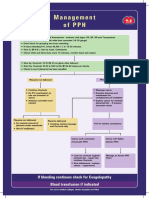
- Management of PPHDocumento1 paginaManagement of PPH098 U.KARTHIK SARAVANA KANTHNessuna valutazione finora
- Warlock of The Magus World - Arc 2Documento808 pagineWarlock of The Magus World - Arc 2Hitsuin MoviesNessuna valutazione finora
- 皮膚病介紹Documento93 pagine皮膚病介紹MK CameraNessuna valutazione finora
- RT-PCR Kit for RNA Detection up to 6.5kbDocumento14 pagineRT-PCR Kit for RNA Detection up to 6.5kbLatifa Putri FajrNessuna valutazione finora
- Advt - R-09-2023 WT ResultDocumento16 pagineAdvt - R-09-2023 WT ResultAvinash Kumar SinghNessuna valutazione finora