Potrebbero piacerti anche
- Mayer 1998 Tetrahedron-LettersDocumento4 pagineMayer 1998 Tetrahedron-LettersEdwar CortesNessuna valutazione finora
- 303 PDFDocumento12 pagine303 PDFSilvanaMedhatNessuna valutazione finora
- Lanthanide Amides ( (Me Si) N) Ln (μ-Cl) Li (THF) Catalyzed Hydrophosphonylation of Aryl AldehydesDocumento4 pagineLanthanide Amides ( (Me Si) N) Ln (μ-Cl) Li (THF) Catalyzed Hydrophosphonylation of Aryl AldehydesDiogomussumNessuna valutazione finora
- An Efficient Synthesis of 5,7-Dimethoxy-4-Methylphthalide, A Key Intermediate in The Synthesis of Mycophenolic AcidDocumento2 pagineAn Efficient Synthesis of 5,7-Dimethoxy-4-Methylphthalide, A Key Intermediate in The Synthesis of Mycophenolic AcidAmeerul HazeeqNessuna valutazione finora
- Two-Step Synthesis of Aza-And Diazaindoles From Chloroamino - N-Heterocycles Using EthoxyvinylborolaneDocumento5 pagineTwo-Step Synthesis of Aza-And Diazaindoles From Chloroamino - N-Heterocycles Using EthoxyvinylborolaneSilvanaMedhatNessuna valutazione finora
- AN IMPROVED SYNTHESIS OF (+) - 2-TROPINONE - Chunming Zhang, Stacey A. Lomenzo, Charles J. Ballay and Mark L. TrudellDocumento2 pagineAN IMPROVED SYNTHESIS OF (+) - 2-TROPINONE - Chunming Zhang, Stacey A. Lomenzo, Charles J. Ballay and Mark L. TrudellTropidinoNessuna valutazione finora
- Microwave-Assisted Synthesis of Dihydropyrimidin-2 (1H) - Ones Using Graphite Supported Lanthanum Chloride As A Mild and e Cient CatalystDocumento3 pagineMicrowave-Assisted Synthesis of Dihydropyrimidin-2 (1H) - Ones Using Graphite Supported Lanthanum Chloride As A Mild and e Cient CatalystnileshsalunkheNessuna valutazione finora
- Nucleosidic Phosphoramidite Synthesis Via PhosphitylationDocumento8 pagineNucleosidic Phosphoramidite Synthesis Via PhosphitylationClarence AG YueNessuna valutazione finora
- 5 Epi LiphagalDocumento3 pagine5 Epi Liphagalglreddy09Nessuna valutazione finora
- David J. Dale Et Al - The Chemical Development of The Commercial Route To Sildenafil: A Case HistoryDocumento6 pagineDavid J. Dale Et Al - The Chemical Development of The Commercial Route To Sildenafil: A Case HistoryRoundSTICNessuna valutazione finora
- Synthesis of Schiff Bases Using Greener MethodologiesDocumento5 pagineSynthesis of Schiff Bases Using Greener MethodologiesDownload_from_ScribdNessuna valutazione finora
- Tetrahedron Letters: Gowravaram Sabitha, K. Purushotham Reddy, S. Purushotham Reddy, J. S. YadavDocumento2 pagineTetrahedron Letters: Gowravaram Sabitha, K. Purushotham Reddy, S. Purushotham Reddy, J. S. Yadavmunnav416Nessuna valutazione finora
- Tetracycline HarvardDocumento2 pagineTetracycline HarvardanisarizcaNessuna valutazione finora
- M. Curini, F. Epifano, S. Genovese, M.C. Marcotullio, O. Rosati. Ytterbium Triflate-Promoted TandemDocumento3 pagineM. Curini, F. Epifano, S. Genovese, M.C. Marcotullio, O. Rosati. Ytterbium Triflate-Promoted TandemMariel MedinaNessuna valutazione finora
- (5C + 1S) Annulation: A Facile and Efficient Synthetic Route Toward Functionalized 2,3-Dihydrothiopyran-4-OnesDocumento4 pagine(5C + 1S) Annulation: A Facile and Efficient Synthetic Route Toward Functionalized 2,3-Dihydrothiopyran-4-OnesRahul BadruNessuna valutazione finora
- 2009 JOC AziridinesDocumento8 pagine2009 JOC AziridinesCarmen SimalNessuna valutazione finora
- Organic Letters (2008), 10 (17), 3907-3909Documento3 pagineOrganic Letters (2008), 10 (17), 3907-3909James TianNessuna valutazione finora
- HecogeninaDocumento4 pagineHecogeninaFernando Torres PérezNessuna valutazione finora
- Modification of The Hofmann Rearrangement, Synthesis of Methyl CarbamatesDocumento3 pagineModification of The Hofmann Rearrangement, Synthesis of Methyl CarbamatesIsmet KutlukNessuna valutazione finora
- Smith Transannular DADocumento4 pagineSmith Transannular DASatyaki MajumdarNessuna valutazione finora
- Enantioselective in AllylationDocumento4 pagineEnantioselective in Allylationredevol7Nessuna valutazione finora
- Synthesis of Analogues of 1,3-Dihydroxyacetone Phosphate and Glyceraldehyde 3-Phosphate For Use in Studies of Fructose-1,6-Diphosphate Aldolase'Documento9 pagineSynthesis of Analogues of 1,3-Dihydroxyacetone Phosphate and Glyceraldehyde 3-Phosphate For Use in Studies of Fructose-1,6-Diphosphate Aldolase'Carlos MRgzNessuna valutazione finora
- DMT - Synthesis.solid Phase - ArticleDocumento4 pagineDMT - Synthesis.solid Phase - ArticleConan MehleNessuna valutazione finora
- Total Synthesis of Quercetin 3 SophorotriosideDocumento4 pagineTotal Synthesis of Quercetin 3 SophorotriosideRiskaNessuna valutazione finora
- 4,7-Diphenylisobenzofuran: A Useful Intermediate For The Construction of Phenyl-Substituted AcenesDocumento11 pagine4,7-Diphenylisobenzofuran: A Useful Intermediate For The Construction of Phenyl-Substituted AcenesUyen Lily HNessuna valutazione finora
- Borohydride IodineDocumento4 pagineBorohydride IodineBandita DattaNessuna valutazione finora
- Total Synthesis of Rapamycin 2Documento2 pagineTotal Synthesis of Rapamycin 2COMPAQSR14Nessuna valutazione finora
- Total Synthesis of Sordaricin: Lewis N. Mander and Regan J. ThomsonDocumento4 pagineTotal Synthesis of Sordaricin: Lewis N. Mander and Regan J. ThomsonOskar Martin OrdoñezNessuna valutazione finora
- CochleamyciDocumento4 pagineCochleamyciOscar Martin OrdoñezNessuna valutazione finora
- Rapid Synthesis of Carbohydrate Derivatives, Including Mimetics of C-Linked Disaccharides and C-Linked Aza Disaccharides, Using The Hetero-Diels - Alder ReactionDocumento9 pagineRapid Synthesis of Carbohydrate Derivatives, Including Mimetics of C-Linked Disaccharides and C-Linked Aza Disaccharides, Using The Hetero-Diels - Alder ReactionDiogomussumNessuna valutazione finora
- Syntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesDocumento14 pagineSyntheses of The Enantiomers of 1-Deoxynojirimycin and 1-Deoxyaltronojirimycin Via Chemo-And Diastereoselective Olefinic Oxidation of Unsaturated AminesDiogo DiasNessuna valutazione finora
- Diazotazione NaNO2 NaHSO3Documento3 pagineDiazotazione NaNO2 NaHSO3leda_prandiNessuna valutazione finora
- An Easy Microwave-Assisted Synthesis of Sulfonamides Directly From Sulfonic AcidsDocumento3 pagineAn Easy Microwave-Assisted Synthesis of Sulfonamides Directly From Sulfonic AcidsVirender KumarNessuna valutazione finora
- A Concise and Stereoselective Synthesis of SqualamineDocumento3 pagineA Concise and Stereoselective Synthesis of SqualamineStella AguirreNessuna valutazione finora
- Amantini 2001Documento4 pagineAmantini 2001AMNessuna valutazione finora
- Twofold CH Functionalization: Palladium-Catalyzed Ortho Arylation of AnilidesDocumento5 pagineTwofold CH Functionalization: Palladium-Catalyzed Ortho Arylation of Anilidesmalala000Nessuna valutazione finora
- Observations On The Deprotection of Pinanediol and Pinacol Boronate Esters Via Fluorinated IntermediatesDocumento4 pagineObservations On The Deprotection of Pinanediol and Pinacol Boronate Esters Via Fluorinated IntermediatesSilvanaMedhatNessuna valutazione finora
- Ullmann Acridine SynthesisDocumento4 pagineUllmann Acridine SynthesisChiến NguyễnNessuna valutazione finora
- Tetrahedron Lett. 2011Documento2 pagineTetrahedron Lett. 2011kasliwalrajeshNessuna valutazione finora
- Riboceine Paper 20 1Documento6 pagineRiboceine Paper 20 1api-257130539Nessuna valutazione finora
- Joctamiflu PDFDocumento8 pagineJoctamiflu PDFFikriansyah Bin IrmanNessuna valutazione finora
- Jo 0503299Documento6 pagineJo 0503299Kyucheol PaikNessuna valutazione finora
- Synthesis of New Pyrimidine Derivatives With Evaluation of Their Anti-Inflammatory and Analgesic ActivitiesDocumento11 pagineSynthesis of New Pyrimidine Derivatives With Evaluation of Their Anti-Inflammatory and Analgesic ActivitiesAmer KasidehNessuna valutazione finora
- Akhlaghinia-2009 (Iodacao de Arenos)Documento6 pagineAkhlaghinia-2009 (Iodacao de Arenos)tenorio pauloNessuna valutazione finora
- Ol 052578 PDocumento4 pagineOl 052578 PChitrangadha ReddyNessuna valutazione finora
- Efficient Synthesis of 3-Cyano-6 - (2-Hydroxyphenyl) Pyridines by Multi-Component Condensations On BeadsDocumento3 pagineEfficient Synthesis of 3-Cyano-6 - (2-Hydroxyphenyl) Pyridines by Multi-Component Condensations On BeadsBandita DattaNessuna valutazione finora
- 1 s2.0 S0014827X01011892 MainDocumento5 pagine1 s2.0 S0014827X01011892 Mainjipir64332Nessuna valutazione finora
- 961 Efficient Method Going From OH To Cle3b0Documento3 pagine961 Efficient Method Going From OH To Cle3b0Wolmir NemitzNessuna valutazione finora
- Ylva E. Bergman, Roger Mulder and Patrick Perlmutter - Total Synthesis of 20-Norsalvinorin A. 1. Preparation of A Key IntermediateDocumento4 pagineYlva E. Bergman, Roger Mulder and Patrick Perlmutter - Total Synthesis of 20-Norsalvinorin A. 1. Preparation of A Key IntermediateBic0000Nessuna valutazione finora
- Efficient Synthesis of 1,5-BenzodiazepinesDocumento3 pagineEfficient Synthesis of 1,5-BenzodiazepinesdoubleffectNessuna valutazione finora
- Joc - 7099Documento8 pagineJoc - 7099Diogo DiasNessuna valutazione finora
- Synthesis of Benzimidazole Using Boric AcidDocumento5 pagineSynthesis of Benzimidazole Using Boric Acidg20kpNessuna valutazione finora
- Flash SublimationDocumento1 paginaFlash SublimationIan OttoNessuna valutazione finora
- Zutter2008 PDFDocumento8 pagineZutter2008 PDFTom ChanNessuna valutazione finora
- Benzyl AlcoholDocumento3 pagineBenzyl AlcohollegolNessuna valutazione finora
- IsoxazolesDocumento7 pagineIsoxazolesiliana56Nessuna valutazione finora
- Molbank: Synthesis and Characterization of A Novel 2-PyrazolineDocumento4 pagineMolbank: Synthesis and Characterization of A Novel 2-PyrazolineAndre BertuahNessuna valutazione finora
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesDa EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNessuna valutazione finora
- Transition Metal Catalyzed Pyrimidine, Pyrazine, Pyridazine and Triazine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesDa EverandTransition Metal Catalyzed Pyrimidine, Pyrazine, Pyridazine and Triazine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNessuna valutazione finora
- Chlorinated Dioxins & Related Compounds: Impact on the EnvironmentDa EverandChlorinated Dioxins & Related Compounds: Impact on the EnvironmentNessuna valutazione finora
- Hello!! BananaDocumento1 paginaHello!! BananaFurphy1Nessuna valutazione finora
- Flocculation and CoagulationDocumento1 paginaFlocculation and CoagulationFurphy10% (1)
- TimeDocumento2 pagineTimeFurphy1Nessuna valutazione finora
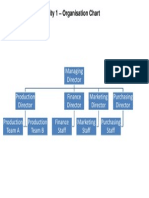
- Activity 1 - Organisation ChartDocumento1 paginaActivity 1 - Organisation ChartFurphy1Nessuna valutazione finora
- SortedDocumento1 paginaSortedFurphy1Nessuna valutazione finora
- SortedDocumento1 paginaSortedFurphy1Nessuna valutazione finora
- Drug Acceptor InteractionsDocumento430 pagineDrug Acceptor InteractionsALNessuna valutazione finora
- ElectrochemistryDocumento62 pagineElectrochemistryRalf ReinhartNessuna valutazione finora
- HalogensDocumento16 pagineHalogensRaju SinghNessuna valutazione finora
- 1 - Chapter 1 PDFDocumento82 pagine1 - Chapter 1 PDFMeenakshi AnilNessuna valutazione finora
- Final Thesis AbDocumento81 pagineFinal Thesis Abfayo100% (1)
- BioplasticDocumento16 pagineBioplasticShubham JaiswalNessuna valutazione finora
- Chem Lab Report 11.doneDocumento14 pagineChem Lab Report 11.donejasnaldNessuna valutazione finora
- CHEMISTRY PROJECT - MergedDocumento14 pagineCHEMISTRY PROJECT - Mergedarpitarathore024Nessuna valutazione finora
- Section ADocumento2 pagineSection AApple KWNessuna valutazione finora
- QOR ChartDocumento2 pagineQOR ChartLoreto StackNessuna valutazione finora
- 20.1999.kinetic and Catalytic Aspects in The Hydrogen Peroxide Production Via AnthraquinoneDocumento8 pagine20.1999.kinetic and Catalytic Aspects in The Hydrogen Peroxide Production Via AnthraquinonesophixNessuna valutazione finora
- Page 1 of 17 Sacred Heart of Mary Girls' SchoolDocumento17 paginePage 1 of 17 Sacred Heart of Mary Girls' SchoolAngeleena ANTONessuna valutazione finora
- 002 Qa 200906Documento14 pagine002 Qa 200906Student ResearchNessuna valutazione finora
- 1.1intro - RCA CleaningDocumento47 pagine1.1intro - RCA CleaningSiddhant BhattacharyaNessuna valutazione finora
- Bio-Lubricant Production From Vegetable Oil and Animal Fat: International UniversityDocumento48 pagineBio-Lubricant Production From Vegetable Oil and Animal Fat: International UniversityKiệtNguyễnNessuna valutazione finora
- Acrylonitrile-Butadiene-Styrene Copolymer (ABS) : Eco-Profiles of The European Plastics IndustryDocumento14 pagineAcrylonitrile-Butadiene-Styrene Copolymer (ABS) : Eco-Profiles of The European Plastics IndustryMary Grace VelitarioNessuna valutazione finora
- Electron Ionization Electron Ionization (EI, Formerly Known As Electron Impact) Is An Ionization MethodDocumento12 pagineElectron Ionization Electron Ionization (EI, Formerly Known As Electron Impact) Is An Ionization MethodDiego RibeiroNessuna valutazione finora
- Chlorosulfuric AcidDocumento2 pagineChlorosulfuric AcidChristine Juliana CiembolonzNessuna valutazione finora
- TEST-17: JEE MainDocumento20 pagineTEST-17: JEE MainRdNessuna valutazione finora
- Kingdom of Saudi Arabia Royal Commission For Jubail and Yanbu Directorate General For Royal Commission at Jubail Jubail Industrial CityDocumento11 pagineKingdom of Saudi Arabia Royal Commission For Jubail and Yanbu Directorate General For Royal Commission at Jubail Jubail Industrial CityJett SorianoNessuna valutazione finora
- HCI Chem H2 Paper 1 Question PaperDocumento17 pagineHCI Chem H2 Paper 1 Question PaperonnoezNessuna valutazione finora
- Aqa Chem1 2002jan PDFDocumento16 pagineAqa Chem1 2002jan PDFholdonpainendsNessuna valutazione finora
- Coefficient of Linear Thermal ExpansiónDocumento28 pagineCoefficient of Linear Thermal ExpansiónalexisvergararangelNessuna valutazione finora
- Ch4502 DMG pdf-2Documento3 pagineCh4502 DMG pdf-2Karan RavalNessuna valutazione finora
- Diffusion Effects in Enzymes Immobilized in A Porous MatrixDocumento18 pagineDiffusion Effects in Enzymes Immobilized in A Porous MatrixAegee0% (1)
- C15 Using Our Resources Student Book AnswersDocumento12 pagineC15 Using Our Resources Student Book AnswersjoeNessuna valutazione finora
- SECTION A (15 Marks) Answer ALL Questions in This SectionDocumento15 pagineSECTION A (15 Marks) Answer ALL Questions in This SectionFazliawati MahayuddinNessuna valutazione finora
- Chemistry of IndigoDocumento2 pagineChemistry of Indigosunny butterNessuna valutazione finora
- Full Length Article: J. Greco-Duarte, E.D. Cavalcanti-Oliveira, J.A.C. Da Silva, R. Fernandez-Lafuente, D.M.G. FreireDocumento10 pagineFull Length Article: J. Greco-Duarte, E.D. Cavalcanti-Oliveira, J.A.C. Da Silva, R. Fernandez-Lafuente, D.M.G. FreireAna Cristina CollaçoNessuna valutazione finora
- Grade Xii (Chemistry) : Aldehydes, Ketones and Carboxylic Acids (Term - 2) : Most Expecting QuestionsDocumento5 pagineGrade Xii (Chemistry) : Aldehydes, Ketones and Carboxylic Acids (Term - 2) : Most Expecting QuestionsSupreeta KhatiwadaNessuna valutazione finora