Potrebbero piacerti anche
- The Yellow House: A Memoir (2019 National Book Award Winner)Da EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Valutazione: 4 su 5 stelle4/5 (98)
- Mitochondrial Disease FactsheetDocumento3 pagineMitochondrial Disease FactsheetNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeDa EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeValutazione: 4 su 5 stelle4/5 (5795)
- Alpers-Huttenlocher FactsheetDocumento2 pagineAlpers-Huttenlocher FactsheetNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Shoe Dog: A Memoir by the Creator of NikeDa EverandShoe Dog: A Memoir by the Creator of NikeValutazione: 4.5 su 5 stelle4.5/5 (537)
- Session Four: HomeworkDocumento1 paginaSession Four: HomeworkNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureDa EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureValutazione: 4.5 su 5 stelle4.5/5 (474)
- Family History CollectionDocumento1 paginaFamily History CollectionNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Grit: The Power of Passion and PerseveranceDa EverandGrit: The Power of Passion and PerseveranceValutazione: 4 su 5 stelle4/5 (588)
- Genomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationDocumento7 pagineGenomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- On Fire: The (Burning) Case for a Green New DealDa EverandOn Fire: The (Burning) Case for a Green New DealValutazione: 4 su 5 stelle4/5 (74)
- Results Interpretation & Application Toolkit: ManagementDocumento1 paginaResults Interpretation & Application Toolkit: ManagementNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryDa EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryValutazione: 3.5 su 5 stelle3.5/5 (231)
- Informed Consent Talking PointsDocumento4 pagineInformed Consent Talking PointsNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceDa EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceValutazione: 4 su 5 stelle4/5 (895)
- Genetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedDocumento1 paginaGenetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Never Split the Difference: Negotiating As If Your Life Depended On ItDa EverandNever Split the Difference: Negotiating As If Your Life Depended On ItValutazione: 4.5 su 5 stelle4.5/5 (838)
- Family History CollectionDocumento1 paginaFamily History CollectionNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- The Little Book of Hygge: Danish Secrets to Happy LivingDa EverandThe Little Book of Hygge: Danish Secrets to Happy LivingValutazione: 3.5 su 5 stelle3.5/5 (400)
- Informed Consent ChecklistDocumento2 pagineInformed Consent ChecklistNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- CMA Provider ResourcesDocumento1 paginaCMA Provider ResourcesNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersDa EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersValutazione: 4.5 su 5 stelle4.5/5 (345)
- Dravet Syndrome FactsheetDocumento1 paginaDravet Syndrome FactsheetNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- HBOC Red Flags ChecklistDocumento1 paginaHBOC Red Flags ChecklistNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- The Unwinding: An Inner History of the New AmericaDa EverandThe Unwinding: An Inner History of the New AmericaValutazione: 4 su 5 stelle4/5 (45)
- HBOC Family History Collection ToolDocumento4 pagineHBOC Family History Collection ToolNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- Team of Rivals: The Political Genius of Abraham LincolnDa EverandTeam of Rivals: The Political Genius of Abraham LincolnValutazione: 4.5 su 5 stelle4.5/5 (234)
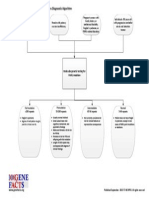
- Fragile X Syndrome Diagnostic AlgorithmDocumento1 paginaFragile X Syndrome Diagnostic AlgorithmNational Coalition for Health Professional Education in GeneticsNessuna valutazione finora
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyDa EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyValutazione: 3.5 su 5 stelle3.5/5 (2259)
- Zoafshan Ms17 Ce ThesisDocumento61 pagineZoafshan Ms17 Ce ThesisJulia MartinNessuna valutazione finora
- Lecture NotesDocumento56 pagineLecture NotesLalaine April E. Ortiola100% (13)
- Title Card Guide Card Activity Card Assessment Card Enrichment Card Tosumitup Reference Card Answer CardDocumento19 pagineTitle Card Guide Card Activity Card Assessment Card Enrichment Card Tosumitup Reference Card Answer CardMyra Roa GasconNessuna valutazione finora
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaDa EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaValutazione: 4.5 su 5 stelle4.5/5 (266)
- Miyagawa Et Al-2015-Human Genome VariationDocumento4 pagineMiyagawa Et Al-2015-Human Genome Variationece142Nessuna valutazione finora
- The Emperor of All Maladies: A Biography of CancerDa EverandThe Emperor of All Maladies: A Biography of CancerValutazione: 4.5 su 5 stelle4.5/5 (271)
- Augmentin DuoDocumento2 pagineAugmentin Duojunaid hashmiNessuna valutazione finora
- Allergy BDS Seminar ReportDocumento21 pagineAllergy BDS Seminar ReportErSandeepVermaNessuna valutazione finora
- Public Health NutritionDocumento58 paginePublic Health NutritionJaime TaylorNessuna valutazione finora
- Radiographic Standard Operating ProtocolsDocumento220 pagineRadiographic Standard Operating Protocolspurnima4uonlyNessuna valutazione finora
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreDa EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreValutazione: 4 su 5 stelle4/5 (1090)
- Module 4 Immediate Kidney Transplant Care 1 1Documento1 paginaModule 4 Immediate Kidney Transplant Care 1 1fouad tabetNessuna valutazione finora
- Update On Binge EatingDocumento12 pagineUpdate On Binge EatingloloasbNessuna valutazione finora
- ECR 2022 ProgrammeDocumento1.126 pagineECR 2022 ProgrammejbmbritoNessuna valutazione finora
- Implant Dentistry - A Rapidly Evolving PracticeDocumento556 pagineImplant Dentistry - A Rapidly Evolving Practicedrescobedocomf67% (3)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Da EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Valutazione: 4.5 su 5 stelle4.5/5 (121)
- Decreased Cardiac OutputDocumento9 pagineDecreased Cardiac OutputChinita Sangbaan75% (4)
- Mastering Your Adult ADHD A Cognitive Behavioral Treatment ProgramDocumento145 pagineMastering Your Adult ADHD A Cognitive Behavioral Treatment ProgramLilia Botnaru96% (28)
- Canadian Occupational Performance Measure (COPM) in Primary Care: A Profile of PracticeDocumento8 pagineCanadian Occupational Performance Measure (COPM) in Primary Care: A Profile of Practiceisabel gomezNessuna valutazione finora
- Leprosy BookDocumento89 pagineLeprosy BookJanardhan Reddy P VNessuna valutazione finora
- Brain Cancer ThesisDocumento7 pagineBrain Cancer ThesisKristen Flores100% (2)
- Nursing Care PlansDocumento16 pagineNursing Care PlansSantos Kyla Patricia T.Nessuna valutazione finora
- Hemostatic Agentshemorrhage ControlDocumento75 pagineHemostatic Agentshemorrhage ControlNasraldeen MohamedNessuna valutazione finora
- Pediatrics Rapid RevisionDocumento72 paginePediatrics Rapid RevisionWorld MedclickzNessuna valutazione finora
- TLE 9 Organic Agri Q1 M5Documento21 pagineTLE 9 Organic Agri Q1 M5Netchie BajeNessuna valutazione finora
- Philippine Orthopedic Center Hospital Services and Times of AvailabilityDocumento2 paginePhilippine Orthopedic Center Hospital Services and Times of AvailabilityJhay GoloNessuna valutazione finora
- Her Body and Other Parties: StoriesDa EverandHer Body and Other Parties: StoriesValutazione: 4 su 5 stelle4/5 (821)
- Albert Et Al. The Efficacy of Systematic Active Conservative Treatment For Patients With Severe Sciatica - AbstractDocumento2 pagineAlbert Et Al. The Efficacy of Systematic Active Conservative Treatment For Patients With Severe Sciatica - AbstractPremanandi Devi DasiNessuna valutazione finora
- 1 Thyroid Profile Free - PO1593405471 707Documento1 pagina1 Thyroid Profile Free - PO1593405471 707TanmoyNessuna valutazione finora
- G.R. No. 165279, June 7, 2011.: Consent Rubi Li vs. SolimanDocumento17 pagineG.R. No. 165279, June 7, 2011.: Consent Rubi Li vs. SolimanJOAHNNA PAULA CORPUZNessuna valutazione finora
- Phocomelia: A Case Study: March 2014Documento3 paginePhocomelia: A Case Study: March 2014Nexi anessaNessuna valutazione finora
- Ref 4Documento13 pagineRef 4Tiago BaraNessuna valutazione finora
- Collective Trauma Summit 2021 - Laurie - Leitch - The Social Resilience ModelDocumento2 pagineCollective Trauma Summit 2021 - Laurie - Leitch - The Social Resilience ModelEmilia LarraondoNessuna valutazione finora
- Zika Virus: Emerging Arboviral Threat To BangladeshDocumento17 pagineZika Virus: Emerging Arboviral Threat To BangladeshlkokodkodNessuna valutazione finora
- Rosuvastatin: Role in Secondary Prevention of Cardiovascular DiseaseDocumento5 pagineRosuvastatin: Role in Secondary Prevention of Cardiovascular Diseasenasir uddinNessuna valutazione finora
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Da EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Valutazione: 3 su 5 stelle3/5 (1)